

Rare coding variants in ten genes confer substantial risk for schizophrenia
- PMID: 35396579
- PMCID: PMC9805802
- DOI: 10.1038/s41586-022-04556-w
Rare coding variants in ten genes confer substantial risk for schizophrenia
Abstract
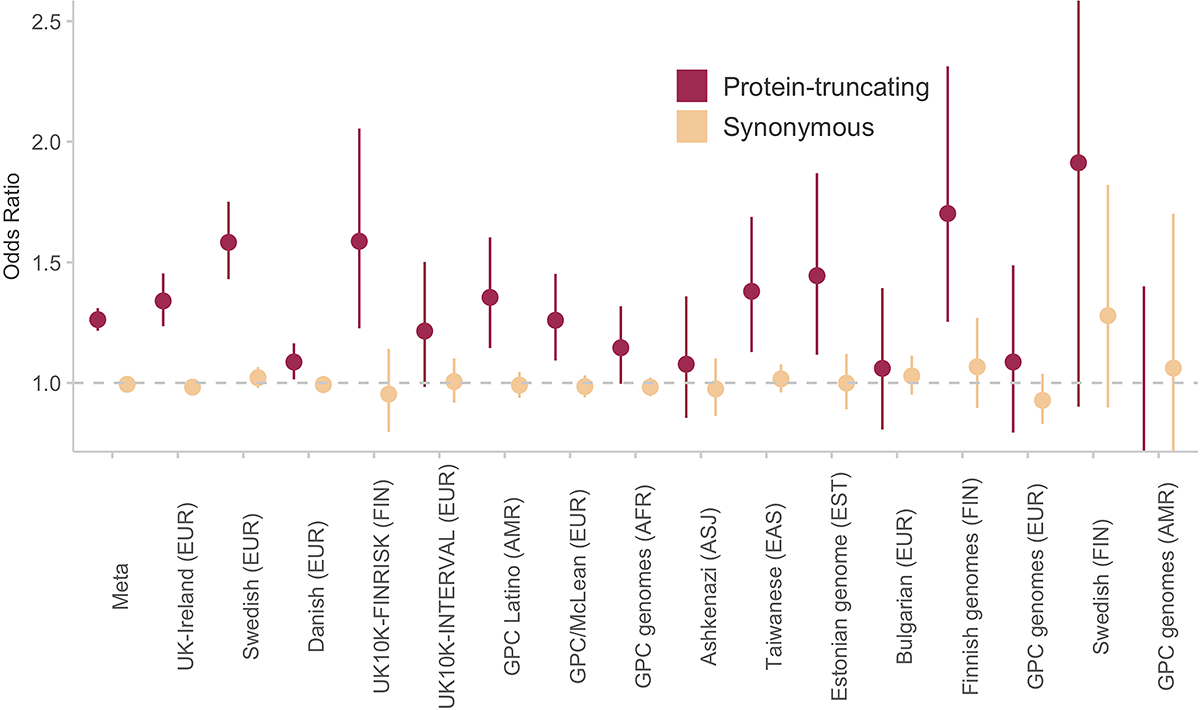
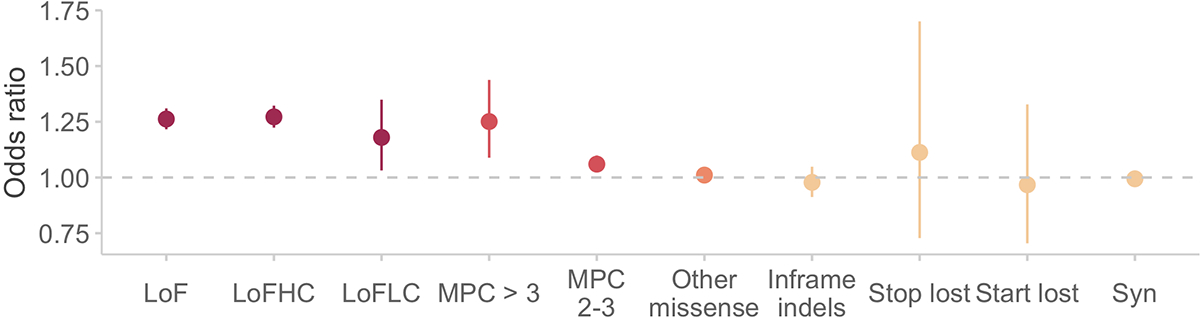
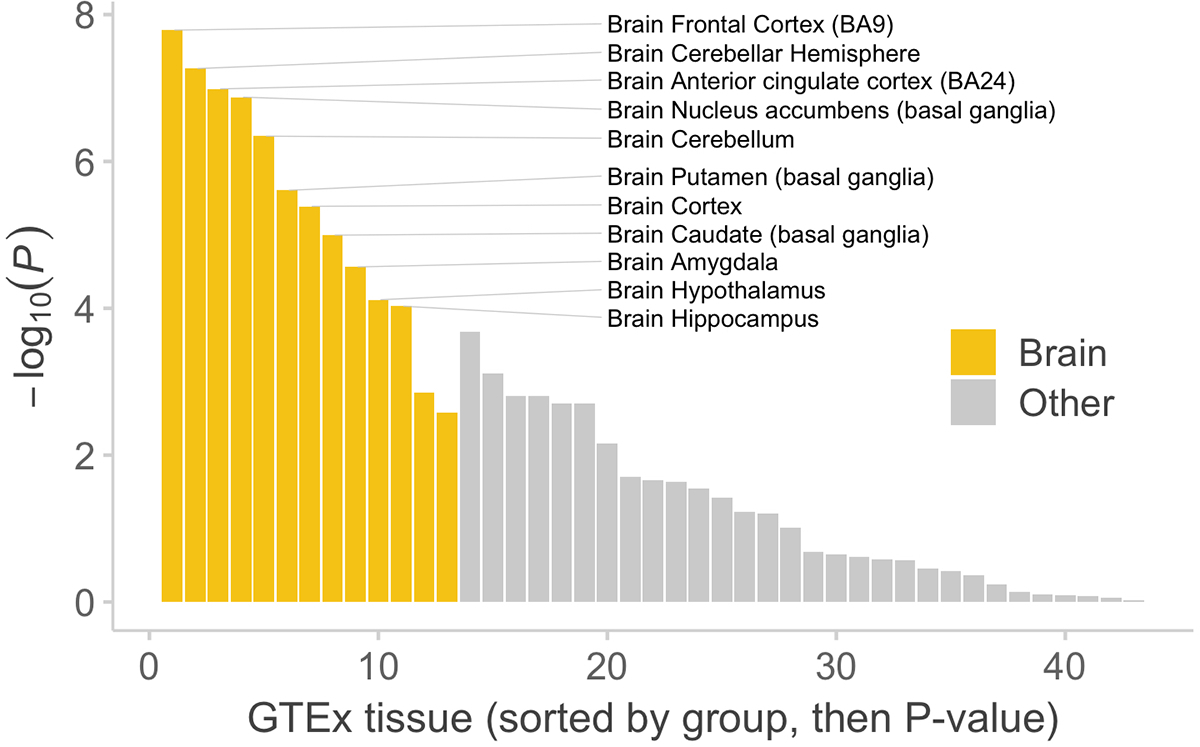
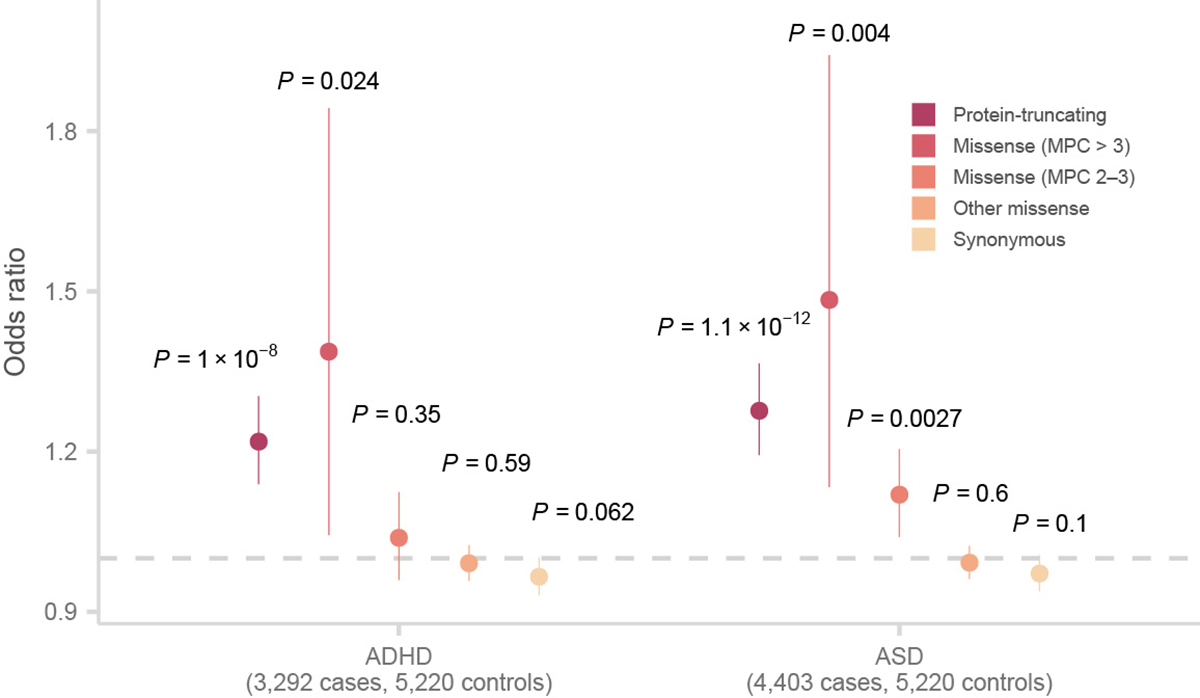
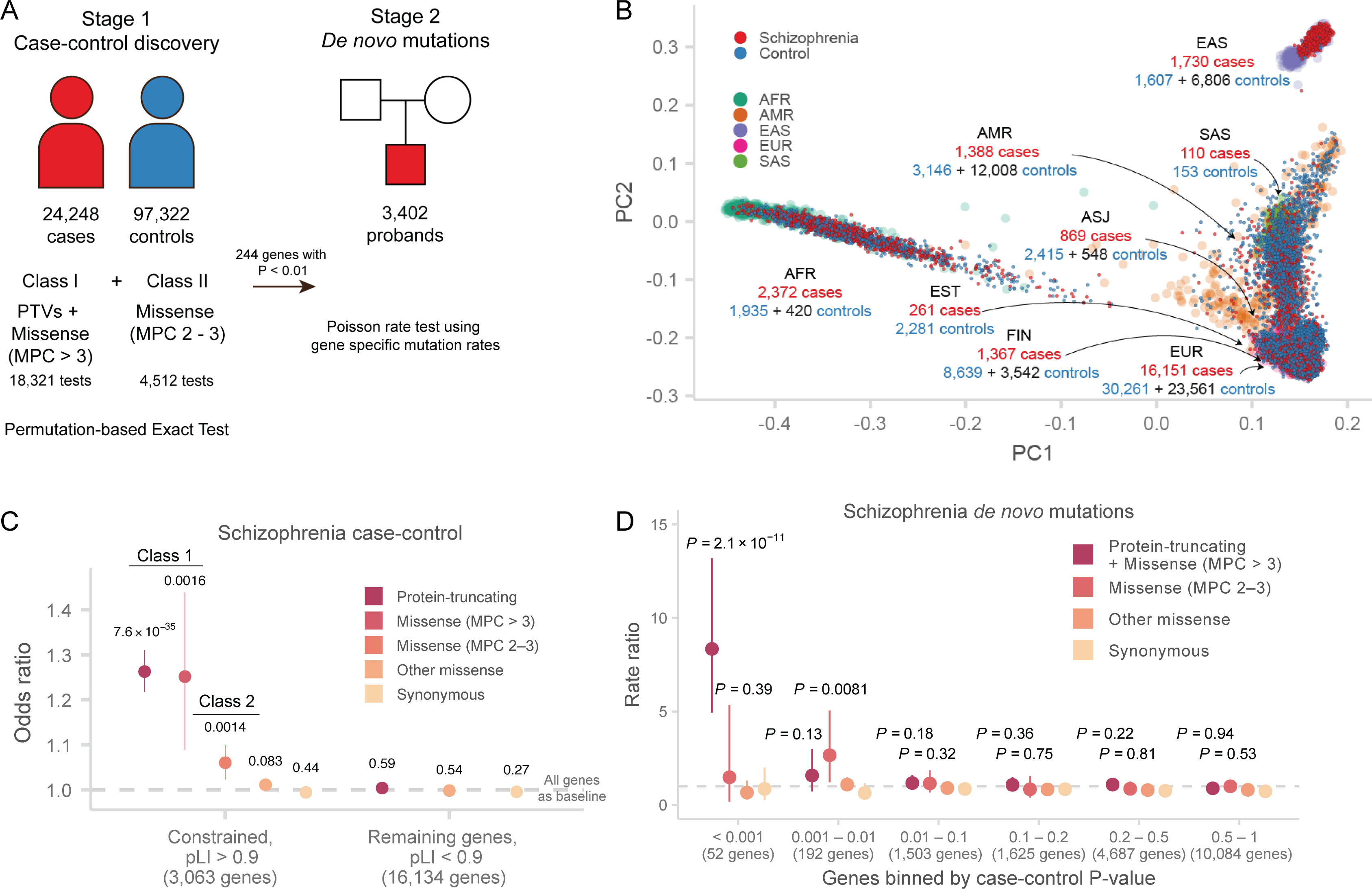
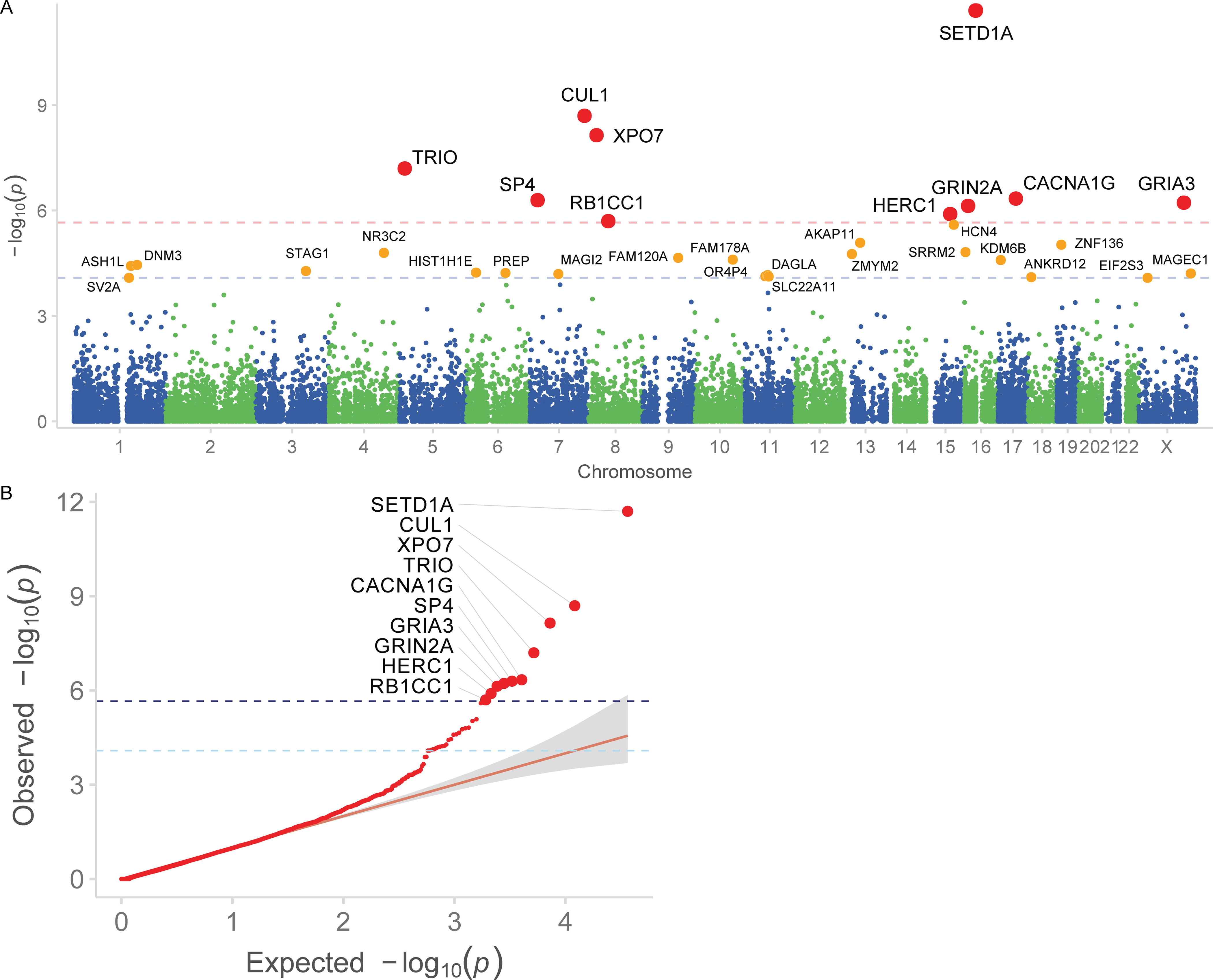
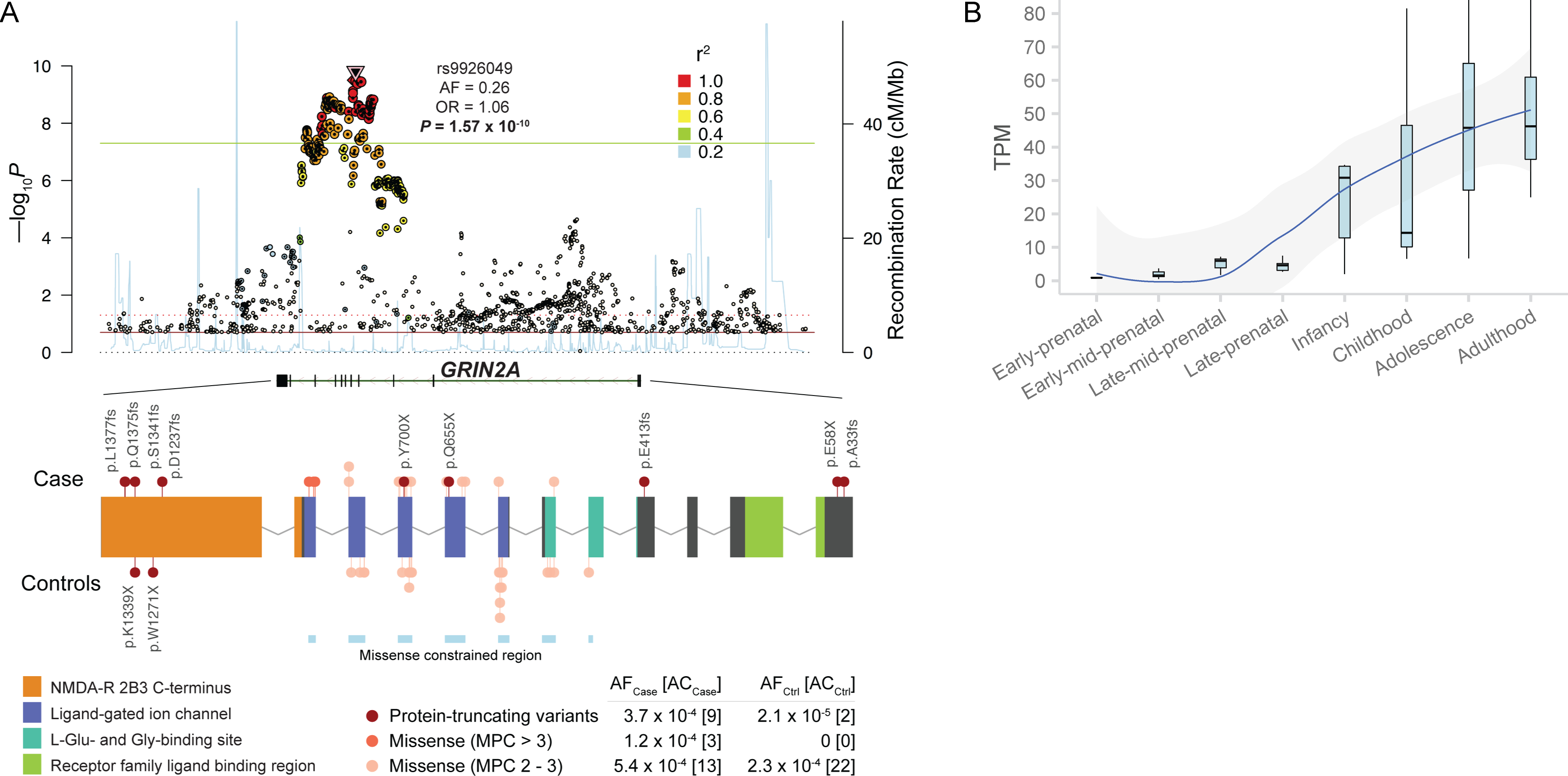
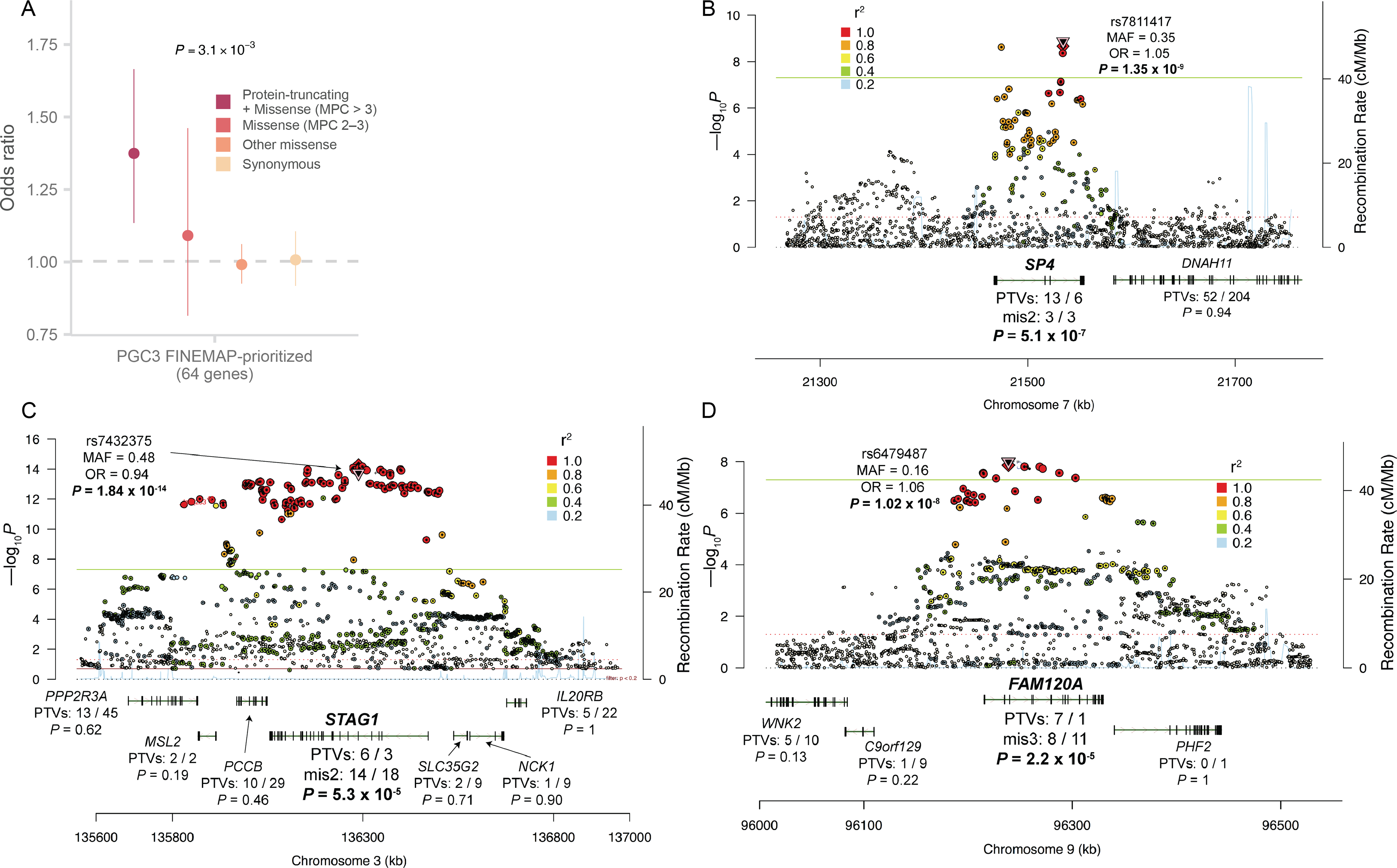
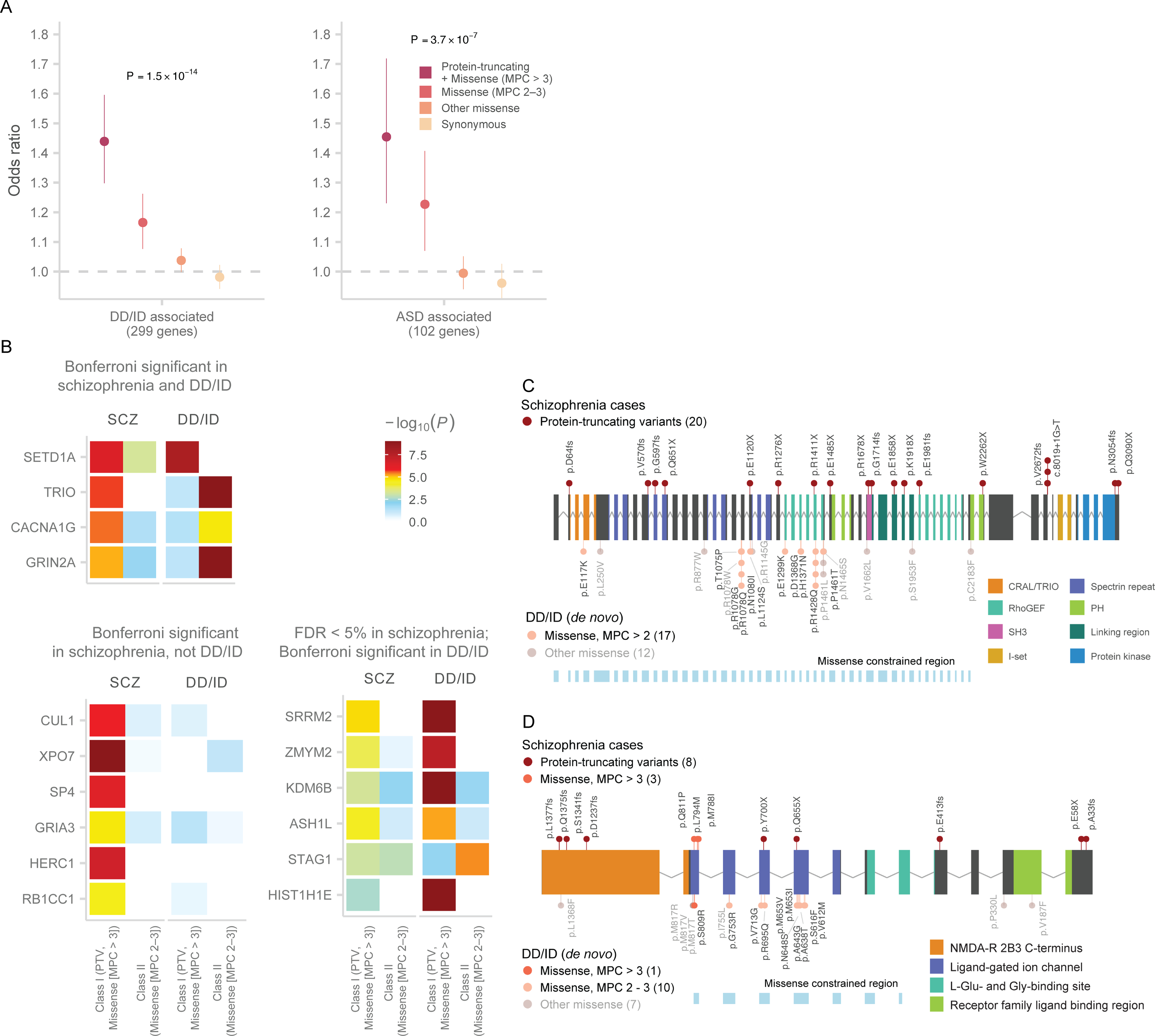
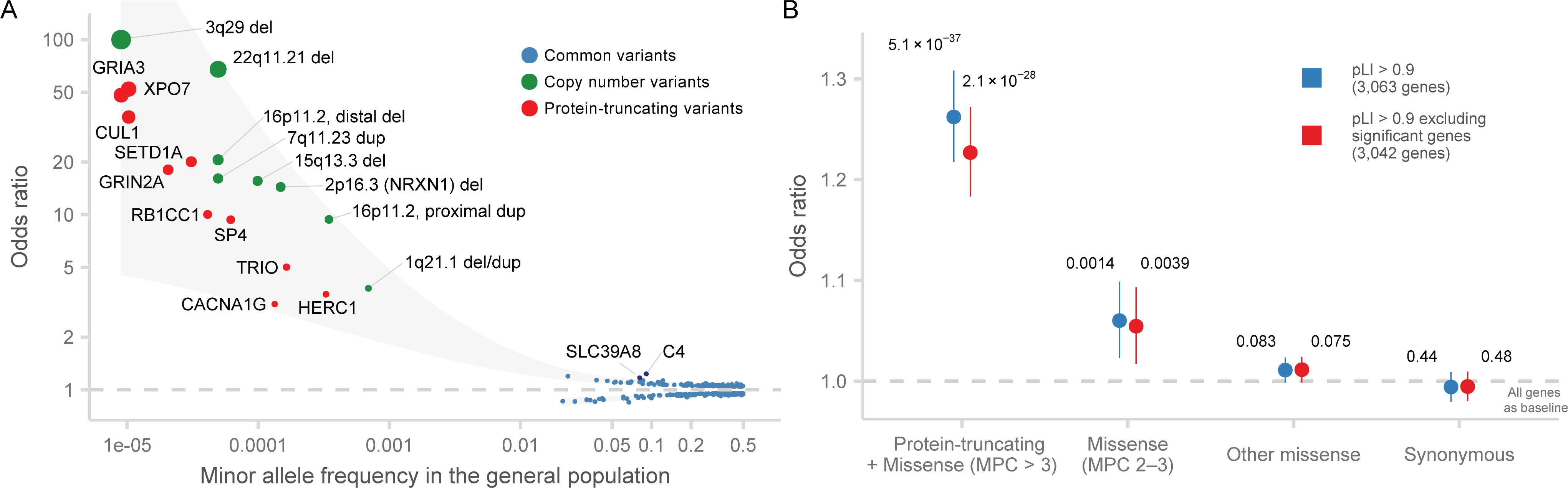
Rare coding variation has historically provided the most direct connections between gene function and disease pathogenesis. By meta-analysing the whole exomes of 24,248 schizophrenia cases and 97,322 controls, we implicate ultra-rare coding variants (URVs) in 10 genes as conferring substantial risk for schizophrenia (odds ratios of 3-50, P < 2.14 × 10-6) and 32 genes at a false discovery rate of <5%. These genes have the greatest expression in central nervous system neurons and have diverse molecular functions that include the formation, structure and function of the synapse. The associations of the NMDA (N-methyl-D-aspartate) receptor subunit GRIN2A and AMPA (α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid) receptor subunit GRIA3 provide support for dysfunction of the glutamatergic system as a mechanistic hypothesis in the pathogenesis of schizophrenia. We observe an overlap of rare variant risk among schizophrenia, autism spectrum disorders1, epilepsy and severe neurodevelopmental disorders2, although different mutation types are implicated in some shared genes. Most genes described here, however, are not implicated in neurodevelopment. We demonstrate that genes prioritized from common variant analyses of schizophrenia are enriched in rare variant risk3, suggesting that common and rare genetic risk factors converge at least partially on the same underlying pathogenic biological processes. Even after excluding significantly associated genes, schizophrenia cases still carry a substantial excess of URVs, which indicates that more risk genes await discovery using this approach.
© 2022. The Author(s), under exclusive licence to Springer Nature Limited.
Conflict of interest statement
Competing Interests
M.J.D. is a founder of Maze Therapeutics and RBNC Therapeutics. B.M.N. is a member of the scientific advisory board at Deep Genomics and RBNC Therapeutics, Member of the scientific advisory committee at Milken and a consultant for Camp4 Therapeutics, Merck and Biogen. A.P. is a member of Astra Zeneca’s Genomics Advisory Board. M.C.O, M.J.O, and J.T.W. are supported by a collaborative research grant from Takeda Pharmaceuticals. D.S.P. was an employee of Genomics plc, all analyses reported in this paper were performed as part of D.S.P.’s employment at the Massachusetts General Hospital and Broad Institute. The remaining authors declare no competing interests.
Figures
Comment in
-
Genetic origins of schizophrenia find common ground.Nature. 2022 Apr;604(7906):433-435. doi: 10.1038/d41586-022-00773-5. Nature. 2022. PMID: 35411108 No abstract available.
References
-
- Schizophrenia Working Group of the Psychiatric Genomics Consortium, Ripke S, Walters JTR & O’Donovan MC Mapping genomic loci prioritises genes and implicates synaptic biology in schizophrenia. medRxiv (2020) doi: 10.1101/2020.09.12.20192922. - DOI
-
- McGrath J, Saha S, Chant D & Welham J Schizophrenia: A Concise Overview of Incidence, Prevalence, and Mortality. Epidemiol. Rev 30, 67–76 (2008). - PubMed
-
- Hjorthøj C, Stürup AE, McGrath JJ & Nordentoft M Years of potential life lost and life expectancy in schizophrenia: a systematic review and meta-analysis. The Lancet Psychiatry 4, 295–301 (2017). - PubMed
MeSH terms
Substances
Grants and funding
- R37 AA009367/AA/NIAAA NIH HHS/United States
- U54 HG003067/HG/NHGRI NIH HHS/United States
- R01 AG064955/AG/NIA NIH HHS/United States
- P50 HD105351/HD/NICHD NIH HHS/United States
- 5R01 AG06495502/NH/NIH HHS/United States
- U01 MH105666/MH/NIMH NIH HHS/United States
- U01 MH105653/MH/NIMH NIH HHS/United States
- U01 DA024417/DA/NIDA NIH HHS/United States
- R01 DA024417/DA/NIDA NIH HHS/United States
- R01 MH085548/MH/NIMH NIH HHS/United States
- R01 MH123451/MH/NIMH NIH HHS/United States
- R01 DA037904/DA/NIDA NIH HHS/United States
- R01 MH094145/MH/NIMH NIH HHS/United States
- R01 MH124851/MH/NIMH NIH HHS/United States
- R01 DA005147/DA/NIDA NIH HHS/United States
- R01 DA036216/DA/NIDA NIH HHS/United States
- R01 AA009367/AA/NIAAA NIH HHS/United States
- R01 MH104964/MH/NIMH NIH HHS/United States
- U01 MH105578/MH/NIMH NIH HHS/United States
- R01 MH116037/MH/NIMH NIH HHS/United States
- U01 MH109539/MH/NIMH NIH HHS/United States
- R01 HG007022/HG/NHGRI NIH HHS/United States
- U01 MH105641/MH/NIMH NIH HHS/United States
- U01 HL117626/HL/NHLBI NIH HHS/United States
- R01 HL117626/HL/NHLBI NIH HHS/United States
- MR/L010305/1/MRC_/Medical Research Council/United Kingdom
- R01 MH085521/MH/NIMH NIH HHS/United States
- R37 DA005147/DA/NIDA NIH HHS/United States
- G0800509/MRC_/Medical Research Council/United Kingdom
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
