

Rapid shear stress-dependent ENaC membrane insertion is mediated by the endothelial glycocalyx and the mineralocorticoid receptor
- PMID: 35397686
- PMCID: PMC8995297
- DOI: 10.1007/s00018-022-04260-y
Rapid shear stress-dependent ENaC membrane insertion is mediated by the endothelial glycocalyx and the mineralocorticoid receptor
Abstract
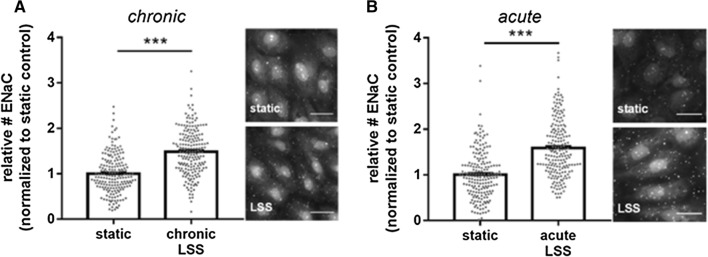
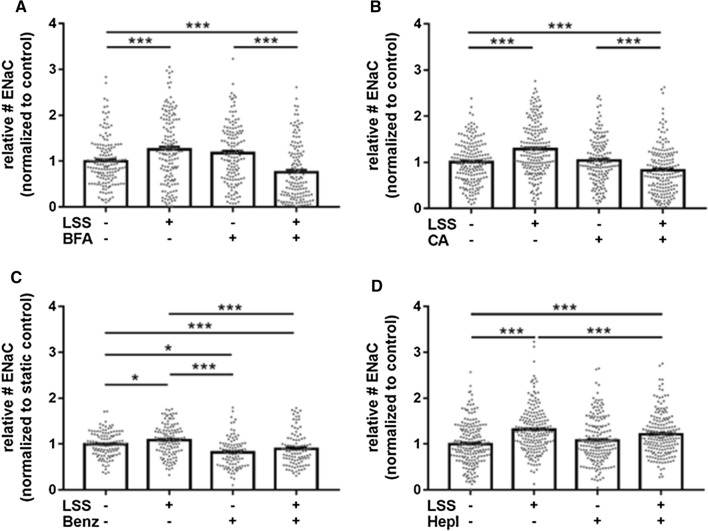
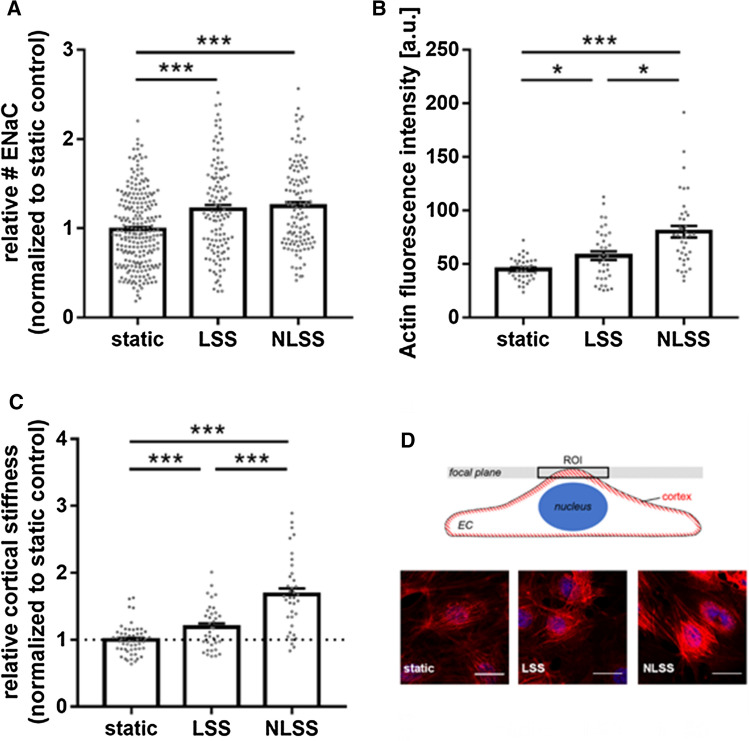
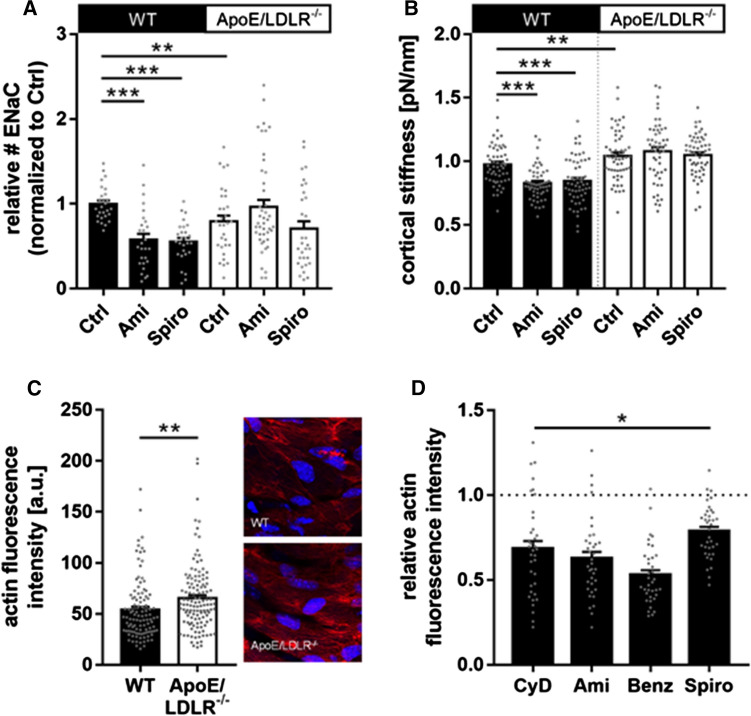
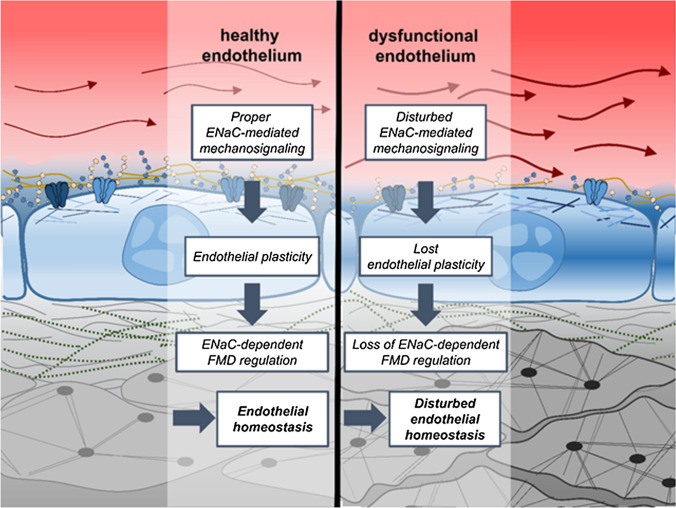
The contribution of the shear stress-sensitive epithelial Na+ channel (ENaC) to the mechanical properties of the endothelial cell surface under (patho)physiological conditions is unclear. This issue was addressed in in vivo and in vitro models for endothelial dysfunction. Cultured human umbilical vein endothelial cells (HUVEC) were exposed to laminar (LSS) or non-laminar shear stress (NLSS). ENaC membrane insertion was quantified using Quantum-dot-based immunofluorescence staining and the mechanical properties of the cell surface were probed with the Atomic Force Microscope (AFM) in vitro and ex vivo in isolated aortae of C57BL/6 and ApoE/LDLR-/- mice. Flow- and acetylcholine-mediated vasodilation was measured in vivo using magnetic resonance imaging. Acute LSS led to a rapid mineralocorticoid receptor (MR)-dependent membrane insertion of ENaC and subsequent stiffening of the endothelial cortex caused by actin polymerization. Of note, NLSS stress further augmented the cortical stiffness of the cells. These effects strongly depend on the presence of the endothelial glycocalyx (eGC) and could be prevented by functional inhibition of ENaC and MR in vitro endothelial cells and ex vivo endothelial cells derived from C57BL/6, but not ApoE/LDLR-/- vessel. In vivo In C57BL/6 vessels, ENaC- and MR inhibition blunted flow- and acetylcholine-mediated vasodilation, while in the dysfunctional ApoE/LDLR-/- vessels, this effect was absent. In conclusion, under physiological conditions, endothelial ENaC, together with the glycocalyx, was identified as an important shear stress sensor and mediator of endothelium-dependent vasodilation. In contrast, in pathophysiological conditions, ENaC-mediated mechanotransduction and endothelium-dependent vasodilation were lost, contributing to sustained endothelial stiffening and dysfunction.
Keywords: ENaC; Endothelial dysfunction; Glycocalyx; Mineralocorticoid receptor; Shear stress.
© 2022. The Author(s).
Conflict of interest statement
Not applicable.
Figures

References
-
- Alexander Y, et al. Endothelial function in cardiovascular medicine: a consensus paper of the European Society of Cardiology Working Groups on Atherosclerosis and Vascular Biology, Aorta and Peripheral Vascular Diseases, Coronary Pathophysiology and Microcirculation, and Thrombosis. Cardiovasc Res. 2021;117:29–42. doi: 10.1093/cvr/cvaa085. - DOI - PMC - PubMed
-
- Chlopicki S, Gryglewski RJ. Angiotensin converting enzyme (ACE) and HydroxyMethylGlutaryl-CoA (HMG-CoA) reductase inhibitors in the forefront of pharmacology of endothelium. Pharmacol Rep. 2005;57:86–96. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
