

A large-scale systematic survey reveals recurring molecular features of public antibody responses to SARS-CoV-2
- PMID: 35397794
- PMCID: PMC8947961
- DOI: 10.1016/j.immuni.2022.03.019
A large-scale systematic survey reveals recurring molecular features of public antibody responses to SARS-CoV-2
Abstract
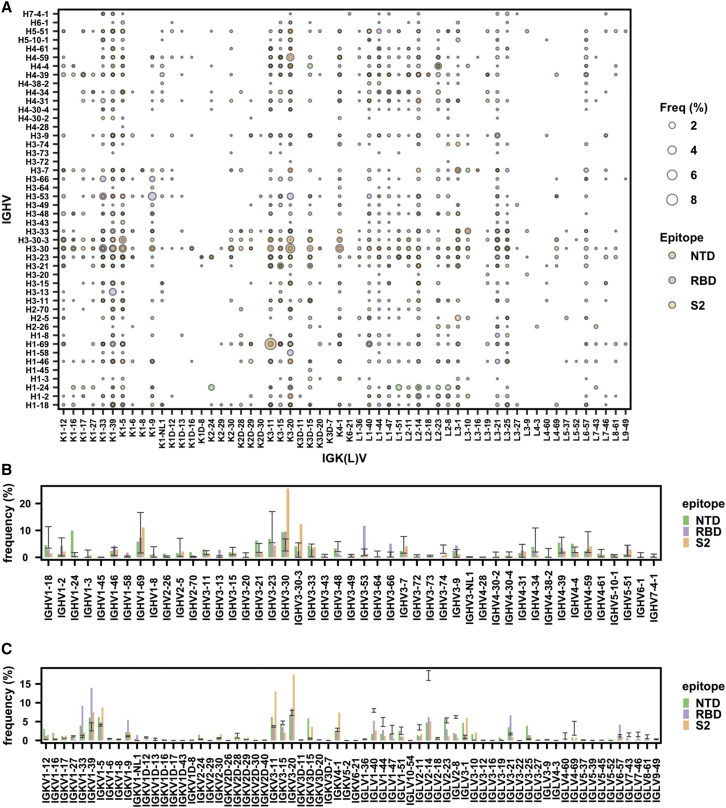
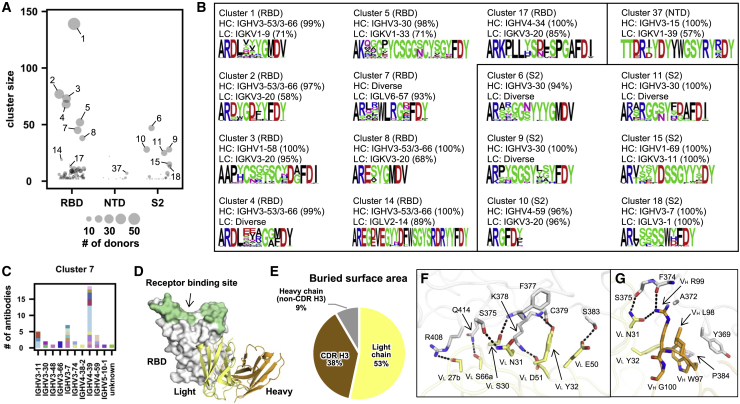
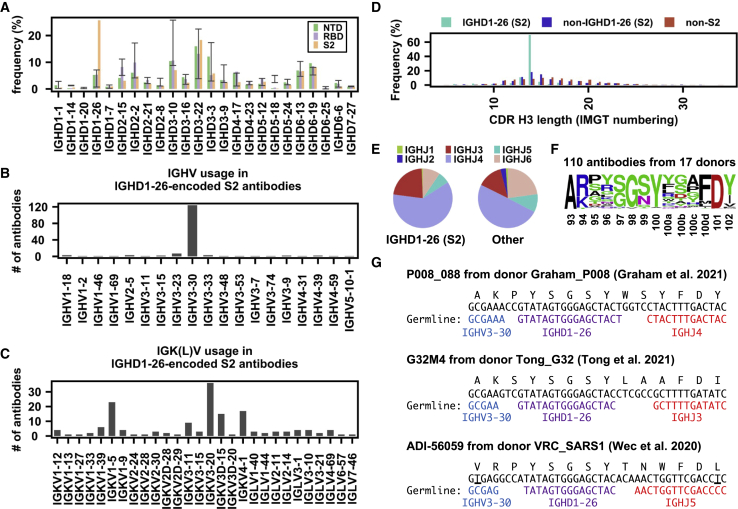
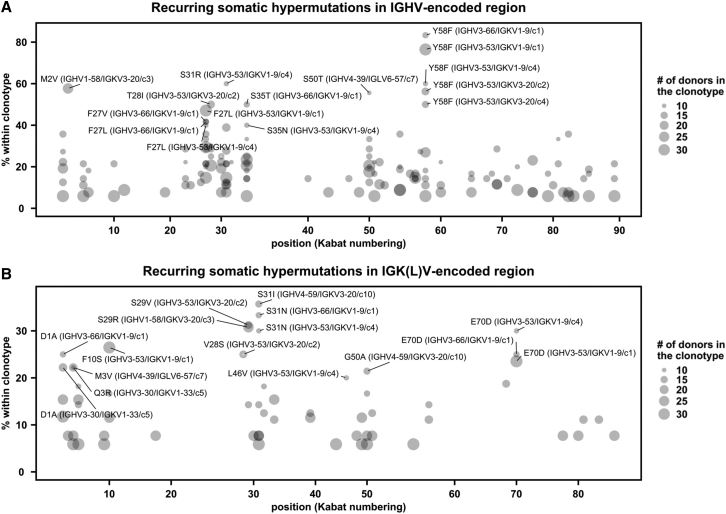
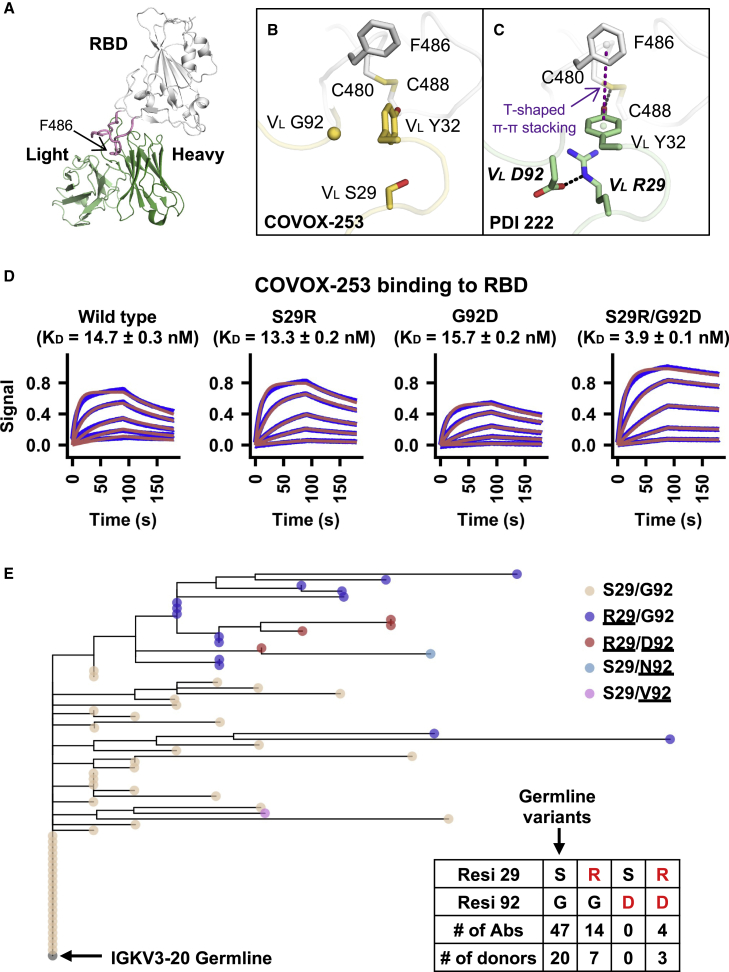
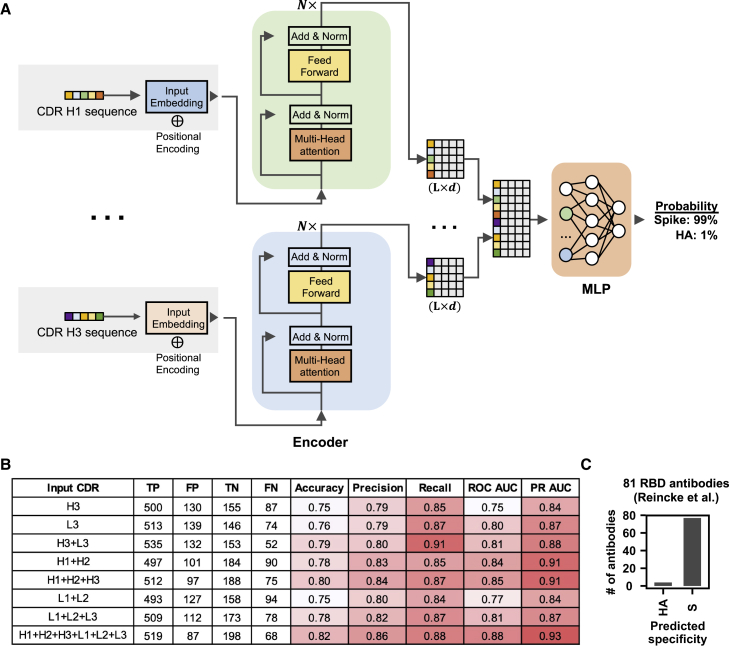
Global research to combat the COVID-19 pandemic has led to the isolation and characterization of thousands of human antibodies to the SARS-CoV-2 spike protein, providing an unprecedented opportunity to study the antibody response to a single antigen. Using the information derived from 88 research publications and 13 patents, we assembled a dataset of ∼8,000 human antibodies to the SARS-CoV-2 spike protein from >200 donors. By analyzing immunoglobulin V and D gene usages, complementarity-determining region H3 sequences, and somatic hypermutations, we demonstrated that the common (public) responses to different domains of the spike protein were quite different. We further used these sequences to train a deep-learning model to accurately distinguish between the human antibodies to SARS-CoV-2 spike protein and those to influenza hemagglutinin protein. Overall, this study provides an informative resource for antibody research and enhances our molecular understanding of public antibody responses.
Keywords: COVID-19; SARS-CoV-2; affinity maturation; antibody; data mining; deep learning; public antibody response; sequence analysis; somatic hypermutation; structural analysis.
Copyright © 2022 The Author(s). Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests The authors declare no competing interests.
Figures
Update of
-
A large-scale systematic survey of SARS-CoV-2 antibodies reveals recurring molecular features.bioRxiv [Preprint]. 2021 Nov 30:2021.11.26.470157. doi: 10.1101/2021.11.26.470157. bioRxiv. 2021. Update in: Immunity. 2022 Jun 14;55(6):1105-1117.e4. doi: 10.1016/j.immuni.2022.03.019. PMID: 34873599 Free PMC article. Updated. Preprint.
References
-
- Abadi M., Barham P., Chen J., Chen Z., Davis A., Dean J., Devin M., Ghemawat S., Irving G., Isard M., et al. 2016. TensorFlow: a system for large-scale machine learning. Paper presented at: Proceedings of the 12th USENIX Symposium on Operating Systems Design and Implementation.
-
- Anderson R.J., Weng Z., Campbell R.K., Jiang X. Main-chain conformational tendencies of amino acids. Proteins. 2005;60:679–689. - PubMed
-
- Andrews S.F., McDermott A.B. Shaping a universally broad antibody response to influenza amidst a variable immunoglobulin landscape. Curr. Opin. Immunol. 2018;53:96–101. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
