

Microglia and Neuroinflammation: Crucial Pathological Mechanisms in Traumatic Brain Injury-Induced Neurodegeneration
- PMID: 35401152
- PMCID: PMC8990307
- DOI: 10.3389/fnagi.2022.825086
Microglia and Neuroinflammation: Crucial Pathological Mechanisms in Traumatic Brain Injury-Induced Neurodegeneration
Abstract
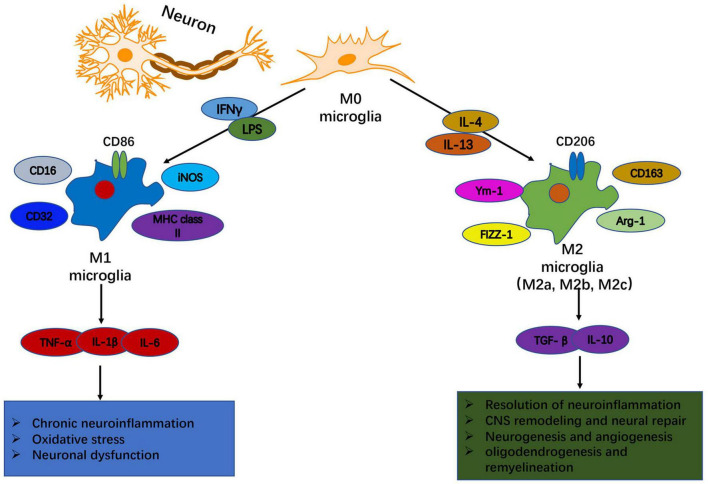
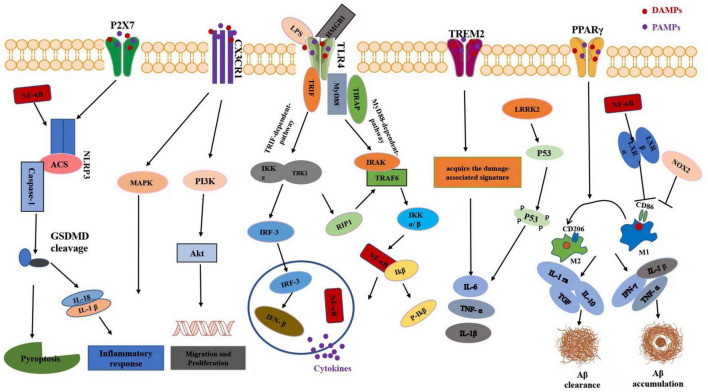
Traumatic brain injury (TBI) is one of the most common diseases in the central nervous system (CNS) with high mortality and morbidity. Patients with TBI usually suffer many sequelae in the life time post injury, including neurodegenerative disorders such as Alzheimer's disease (AD) and Parkinson's disease (PD). However, the pathological mechanisms connecting these two processes have not yet been fully elucidated. It is important to further investigate the pathophysiological mechanisms underlying TBI and TBI-induced neurodegeneration, which will promote the development of precise treatment target for these notorious neurodegenerative consequences after TBI. A growing body of evidence shows that neuroinflammation is a pivotal pathological process underlying chronic neurodegeneration following TBI. Microglia, as the immune cells in the CNS, play crucial roles in neuroinflammation and many other CNS diseases. Of interest, microglial activation and functional alteration has been proposed as key mediators in the evolution of chronic neurodegenerative pathology following TBI. Here, we review the updated studies involving phenotypical and functional alterations of microglia in neurodegeneration after injury, survey key molecules regulating the activities and functional responses of microglia in TBI pathology, and explore their potential implications to chronic neurodegeneration after injury. The work will give us a comprehensive understanding of mechanisms driving TBI-related neurodegeneration and offer novel ideas of developing corresponding prevention and treatment strategies for this disease.
Keywords: central nervous system; microglia; neurodegeneration; neuroinflammation; traumatic brain injury.
Copyright © 2022 Shao, Wang, Wu, Wu and Zhang.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
References
Publication types
LinkOut - more resources
Full Text Sources