

Molecular mechanisms underlying the emergence of polygenetic antifungal drug resistance in msh2 mismatch repair mutants of Cryptococcus
- PMID: 35402912
- PMCID: PMC8986524
- DOI: 10.1093/jacamr/dlac033
Molecular mechanisms underlying the emergence of polygenetic antifungal drug resistance in msh2 mismatch repair mutants of Cryptococcus
Abstract
Background: Fungal infections are common life-threatening diseases amongst immunodeficient individuals. Invasive fungal disease is commonly treated with an azole antifungal agent, resulting in selection pressure and the emergence of drug resistance. Antifungal resistance is associated with higher mortality rates and treatment failure, making the current clinical management of fungal disease very challenging. Clinical isolates from a variety of fungi have been shown to contain mutations in the MSH2 gene, encoding a component of the DNA mismatch repair pathway. Mutation of MSH2 results in an elevated mutation rate that can increase the opportunity for selectively advantageous mutations to occur, accelerating the development of antifungal resistance.
Objectives: To characterize the molecular mechanisms causing the microevolutionary emergence of antifungal resistance in msh2 mismatch repair mutants of Cryptococcus neoformans.
Methods: The mechanisms resulting in the emergence of antifungal resistance were investigated using WGS, characterization of deletion mutants and measuring ploidy changes.
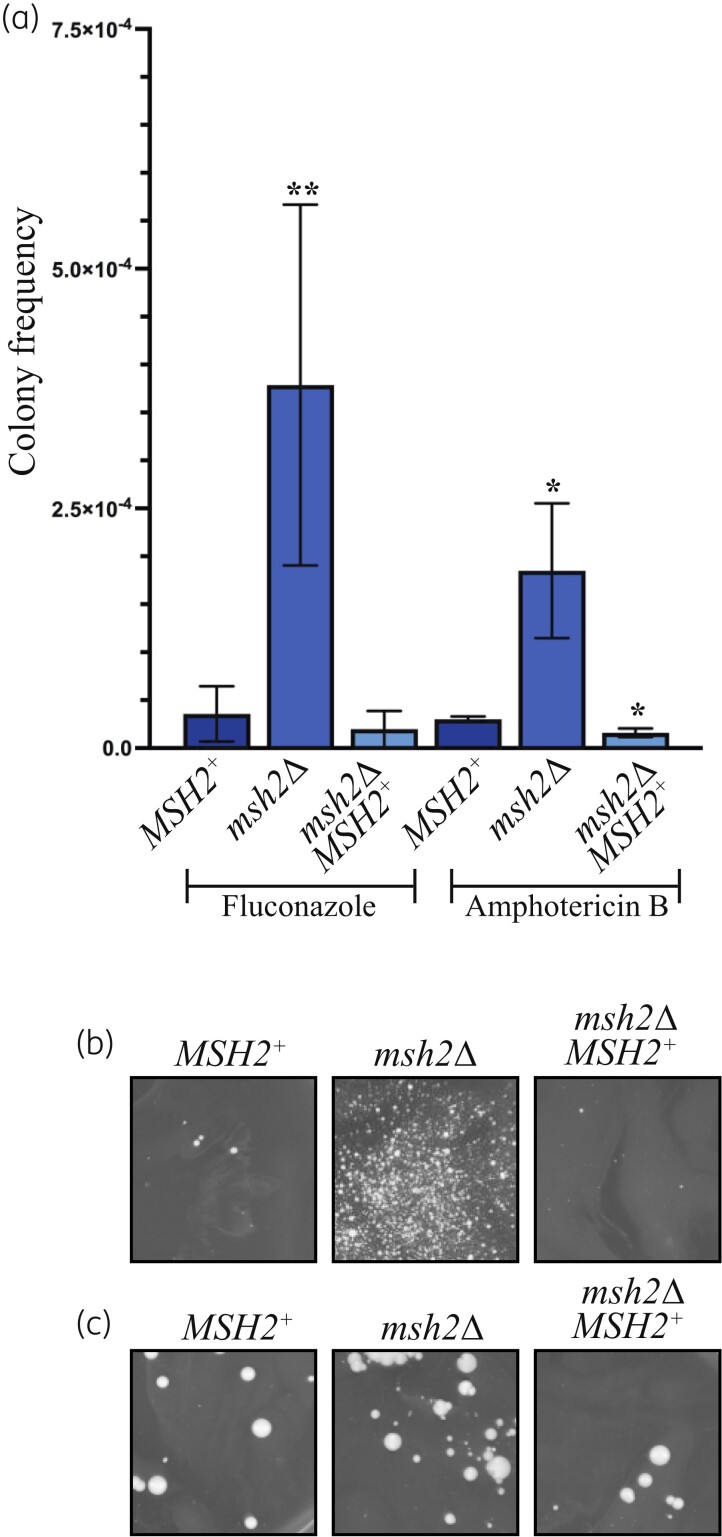
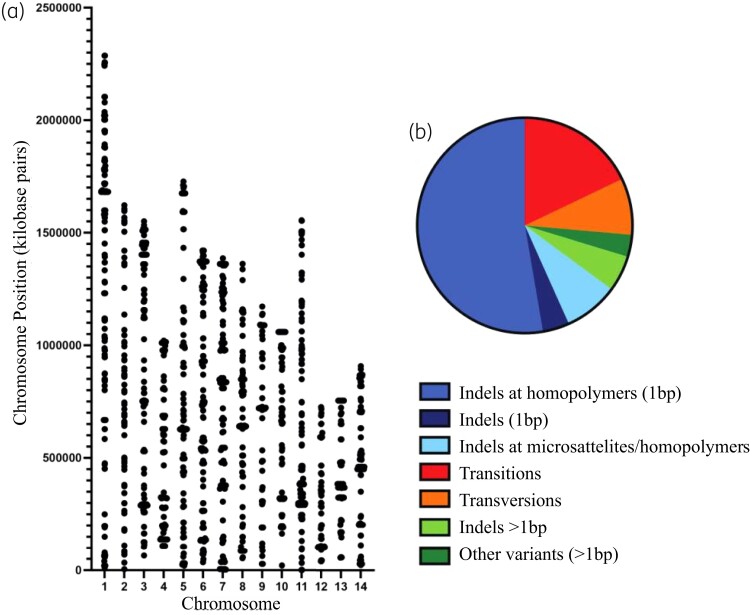
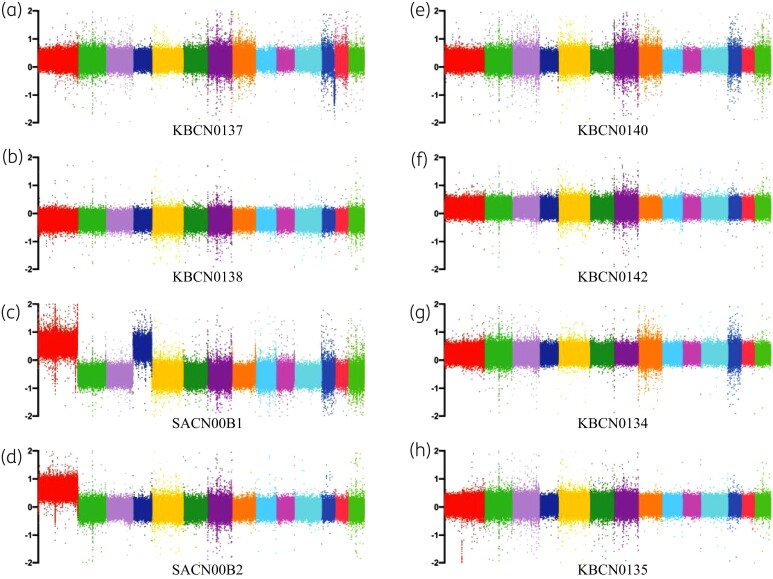
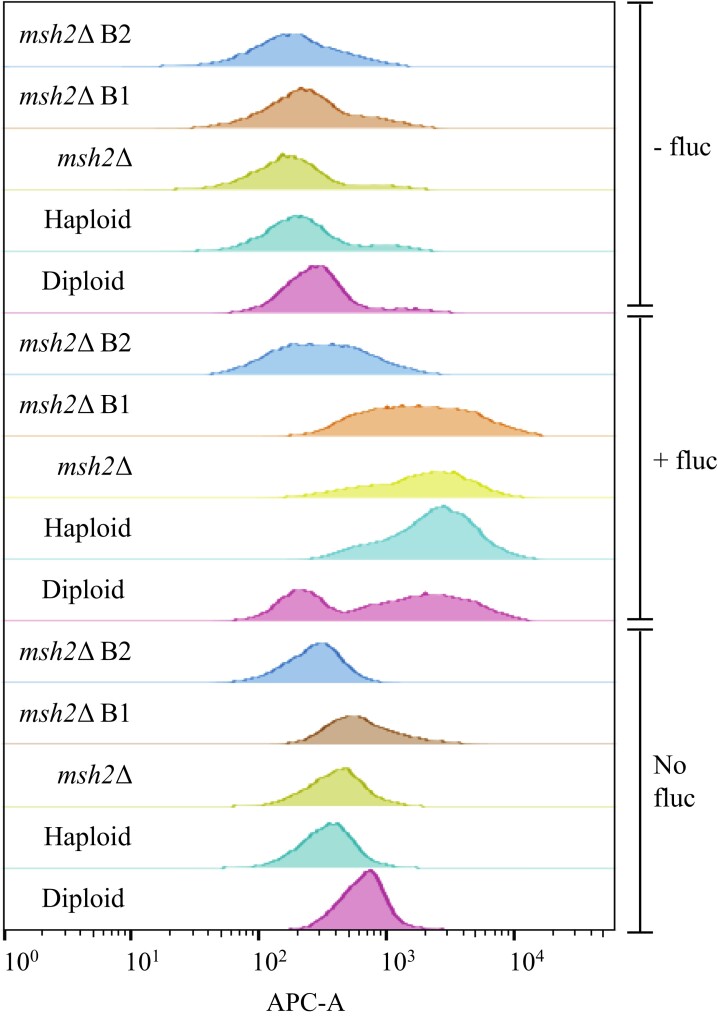
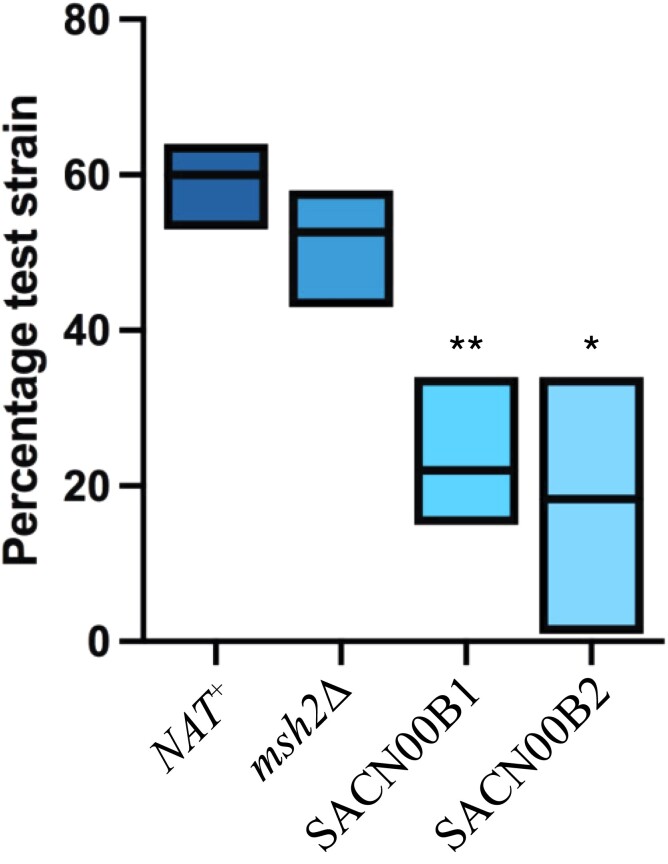
Results: The genomes of resistant strains did not possess mutations in ERG11 or other genes of the ergosterol biosynthesis pathway. Antifungal resistance was due to small contributions from mutations in many genes. MSH2 does not directly affect ploidy changes.
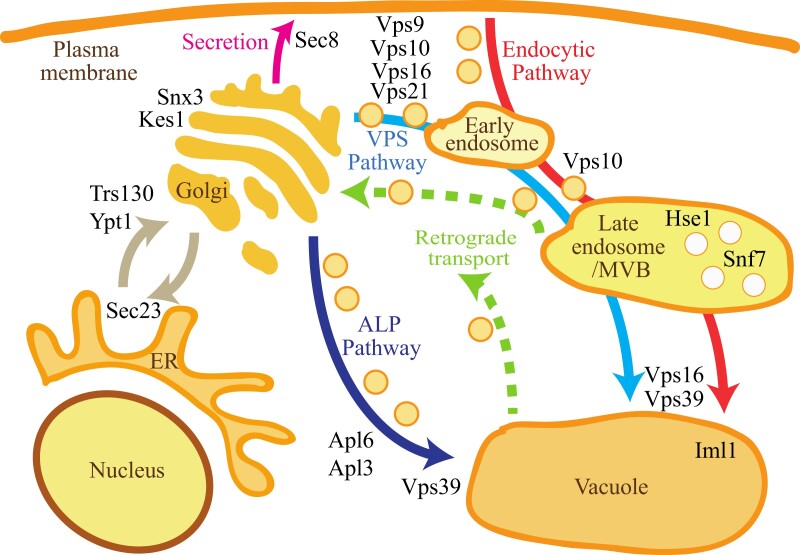
Conclusions: This study provides evidence that resistance to fluconazole can evolve independently of ERG11 mutations. A common microevolutionary route to the emergence of antifungal resistance involves the accumulation of mutations that alter stress signalling, cellular efflux, membrane trafficking, epigenetic modification and aneuploidy. This complex pattern of microevolution highlights the significant challenges posed both to diagnosis and treatment of drug-resistant fungal pathogens.
© The Author(s) 2022. Published by Oxford University Press on behalf of British Society for Antimicrobial Chemotherapy.
Figures
Similar articles
-
Mismatch Repair of DNA Replication Errors Contributes to Microevolution in the Pathogenic Fungus Cryptococcus neoformans.mBio. 2017 May 30;8(3):e00595-17. doi: 10.1128/mBio.00595-17. mBio. 2017. PMID: 28559486 Free PMC article.
-
Resistance in human pathogenic yeasts and filamentous fungi: prevalence, underlying molecular mechanisms and link to the use of antifungals in humans and the environment.Dan Med J. 2016 Oct;63(10):B5288. Dan Med J. 2016. PMID: 27697142 Review.
-
Absence of Azole or Echinocandin Resistance in Candida glabrata Isolates in India despite Background Prevalence of Strains with Defects in the DNA Mismatch Repair Pathway.Antimicrob Agents Chemother. 2018 May 25;62(6):e00195-18. doi: 10.1128/AAC.00195-18. Print 2018 Jun. Antimicrob Agents Chemother. 2018. PMID: 29610199 Free PMC article.
-
The Aspergillus fumigatus Mismatch Repair MSH2 Homolog Is Important for Virulence and Azole Resistance.mSphere. 2019 Aug 7;4(4):e00416-19. doi: 10.1128/mSphere.00416-19. mSphere. 2019. PMID: 31391280 Free PMC article.
-
Antifungal drug resistance in pathogenic fungi.Med Mycol. 1998;36 Suppl 1:119-28. Med Mycol. 1998. PMID: 9988500 Review.
Cited by
-
Combating increased antifungal drug resistance in Cryptococcus, what should we do in the future?Acta Biochim Biophys Sin (Shanghai). 2023 Feb 22;55(4):540-547. doi: 10.3724/abbs.2023011. Acta Biochim Biophys Sin (Shanghai). 2023. PMID: 36815374 Free PMC article. Review.
-
The Microevolution of Antifungal Drug Resistance in Pathogenic Fungi.Microorganisms. 2023 Nov 13;11(11):2757. doi: 10.3390/microorganisms11112757. Microorganisms. 2023. PMID: 38004768 Free PMC article. Review.
-
Cryptococcosis in Southern China: Insights from a Six-Year Retrospective Study in Eastern Guangdong.Infect Drug Resist. 2023 Jul 6;16:4409-4419. doi: 10.2147/IDR.S417968. eCollection 2023. Infect Drug Resist. 2023. PMID: 37435235 Free PMC article.
-
Epigenetic Regulation of Antifungal Drug Resistance.J Fungi (Basel). 2022 Aug 19;8(8):875. doi: 10.3390/jof8080875. J Fungi (Basel). 2022. PMID: 36012862 Free PMC article. Review.
-
CryptoCEN: A Co-Expression Network for Cryptococcus neoformans reveals novel proteins involved in DNA damage repair.bioRxiv [Preprint]. 2023 Aug 18:2023.08.17.553567. doi: 10.1101/2023.08.17.553567. bioRxiv. 2023. Update in: PLoS Genet. 2024 Feb 15;20(2):e1011158. doi: 10.1371/journal.pgen.1011158. PMID: 37645941 Free PMC article. Updated. Preprint.
References
LinkOut - more resources
Full Text Sources