

Molecular pathogenesis of acetaminophen-induced liver injury and its treatment options
- PMID: 35403383
- PMCID: PMC9002247
- DOI: 10.1631/jzus.B2100977
Molecular pathogenesis of acetaminophen-induced liver injury and its treatment options
Abstract
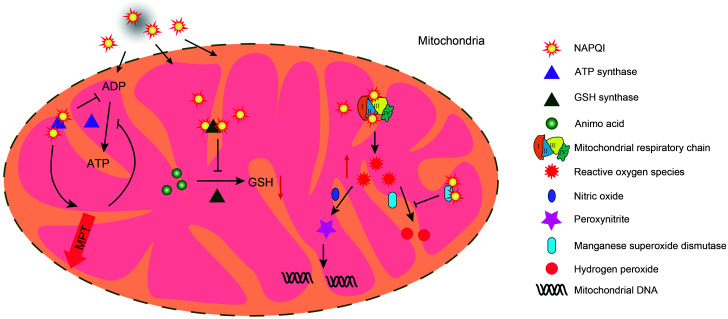
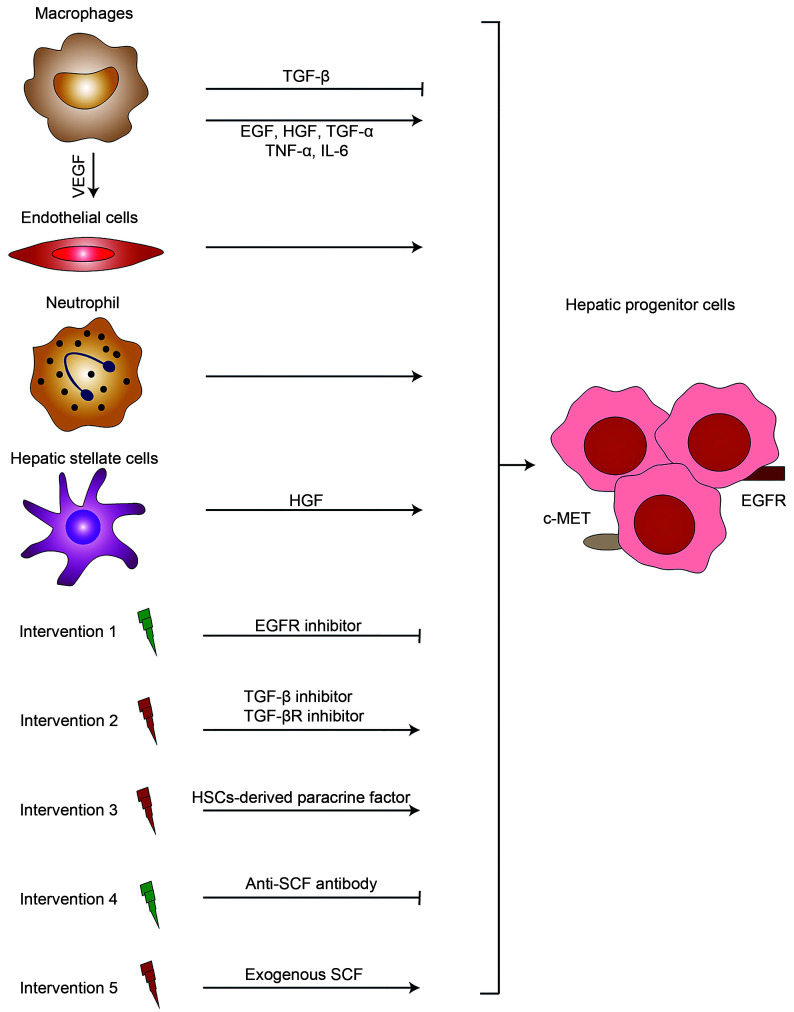
Acetaminophen, also known as N-acetyl-p-aminophenol (APAP), is commonly used as an antipyretic and analgesic agent. APAP overdose can induce hepatic toxicity, known as acetaminophen-induced liver injury (AILI). However, therapeutic doses of APAP can also induce AILI in patients with excessive alcohol intake or who are fasting. Hence, there is a need to understand the potential pathological mechanisms underlying AILI. In this review, we summarize three main mechanisms involved in the pathogenesis of AILI: hepatocyte necrosis, sterile inflammation, and hepatocyte regeneration. The relevant factors are elucidated and discussed. For instance, N-acetyl-p-benzoquinone imine (NAPQI) protein adducts trigger mitochondrial oxidative/nitrosative stress during hepatocyte necrosis, danger-associated molecular patterns (DAMPs) are released to elicit sterile inflammation, and certain growth factors contribute to liver regeneration. Finally, we describe the current potential treatment options for AILI patients and promising novel strategies available to researchers and pharmacists. This review provides a clearer understanding of AILI-related mechanisms to guide drug screening and selection for the clinical treatment of AILI patients in the future.
Keywords: Acetaminophen; Acetaminophen-induced liver injury; Hepatocyte necrosis; Hepatocyte regeneration; Sterile inflammation.
Figures



References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
