

Evolution of binding preferences among whole-genome duplicated transcription factors
- PMID: 35404235
- PMCID: PMC9000951
- DOI: 10.7554/eLife.73225
Evolution of binding preferences among whole-genome duplicated transcription factors
Abstract
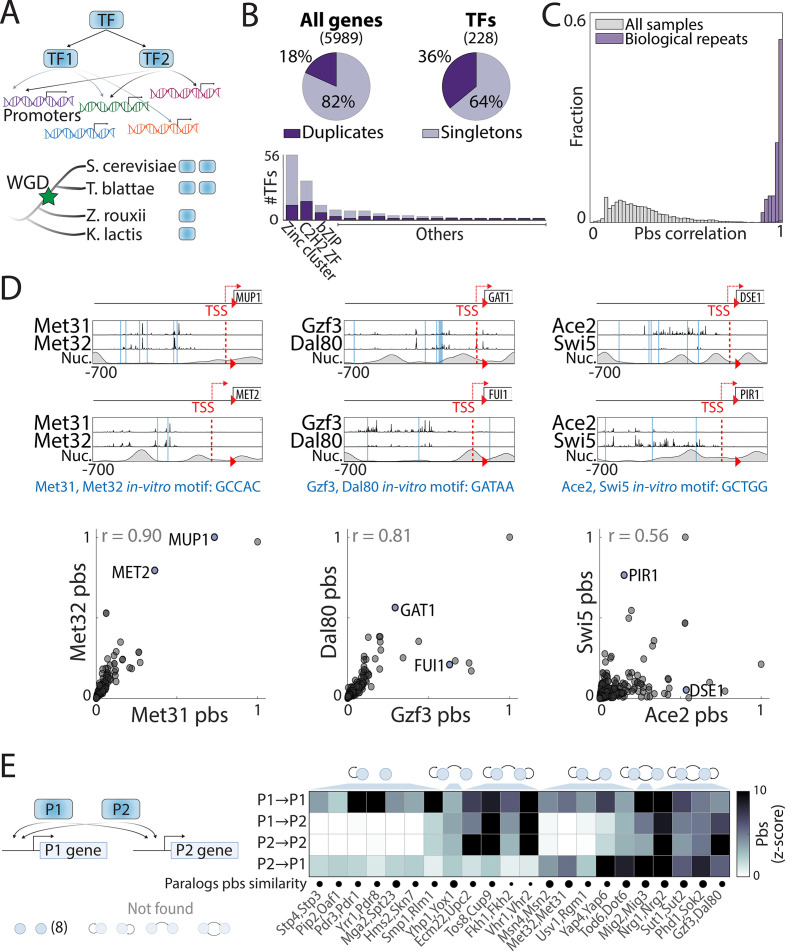
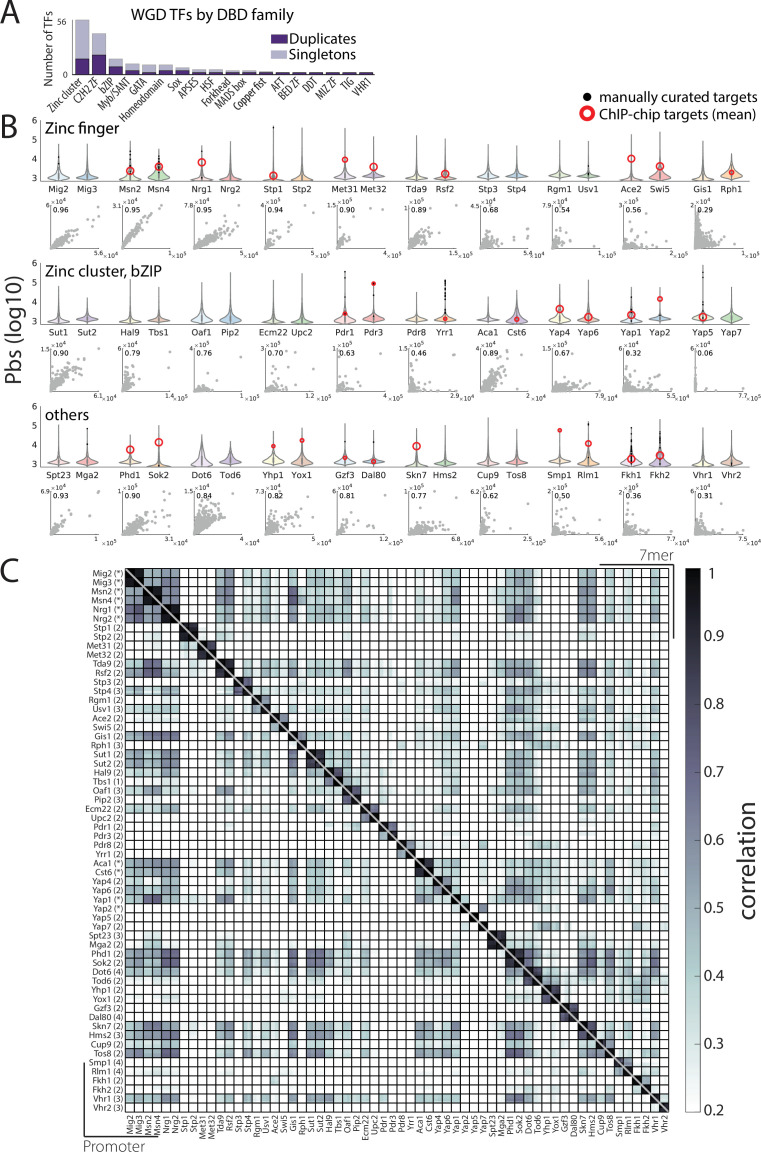
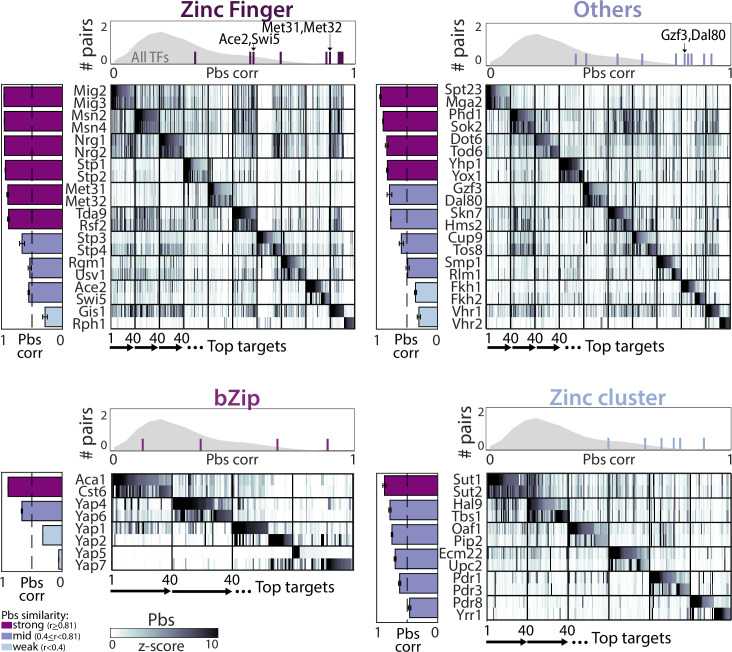
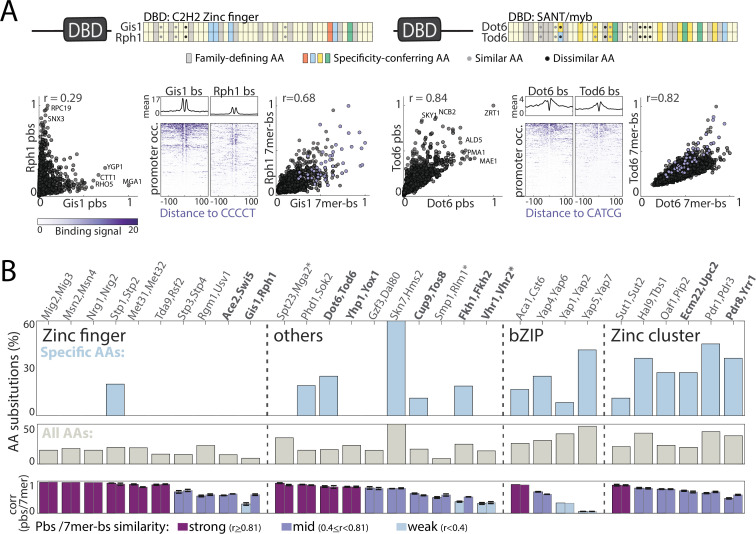
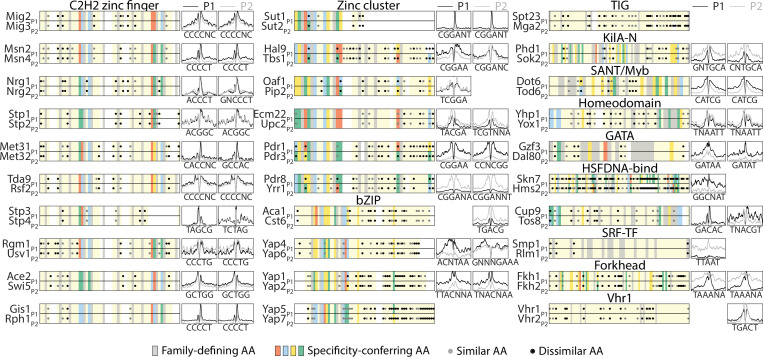
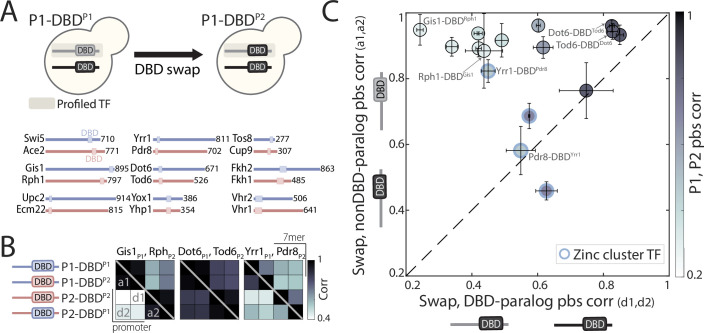
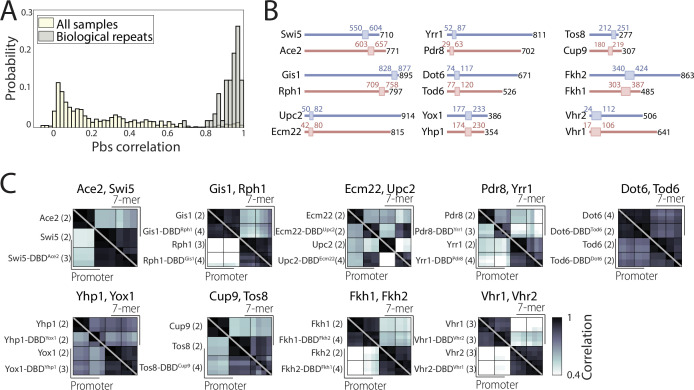
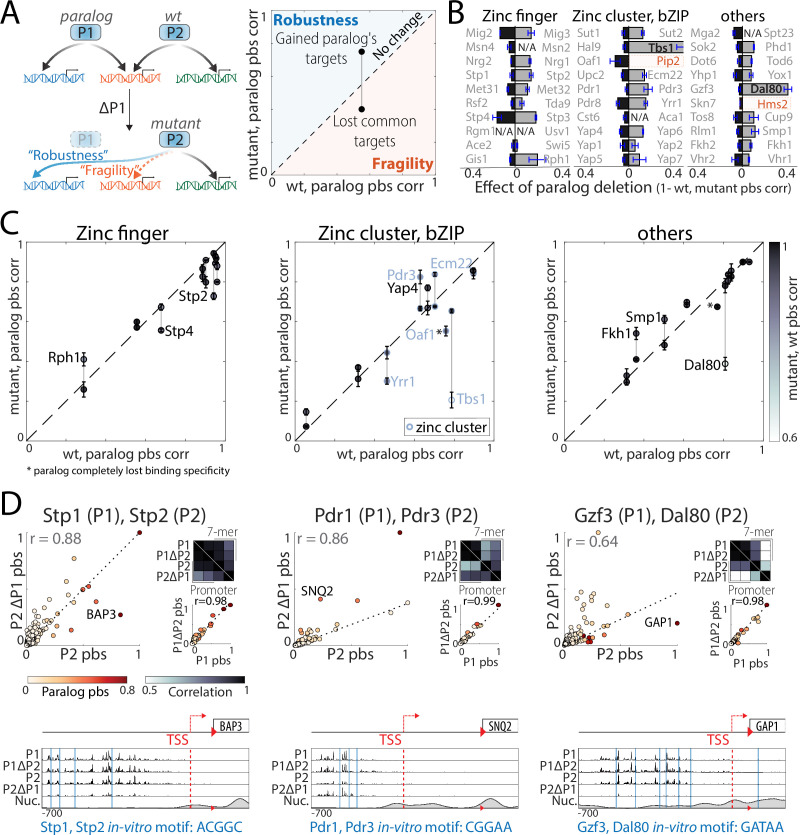
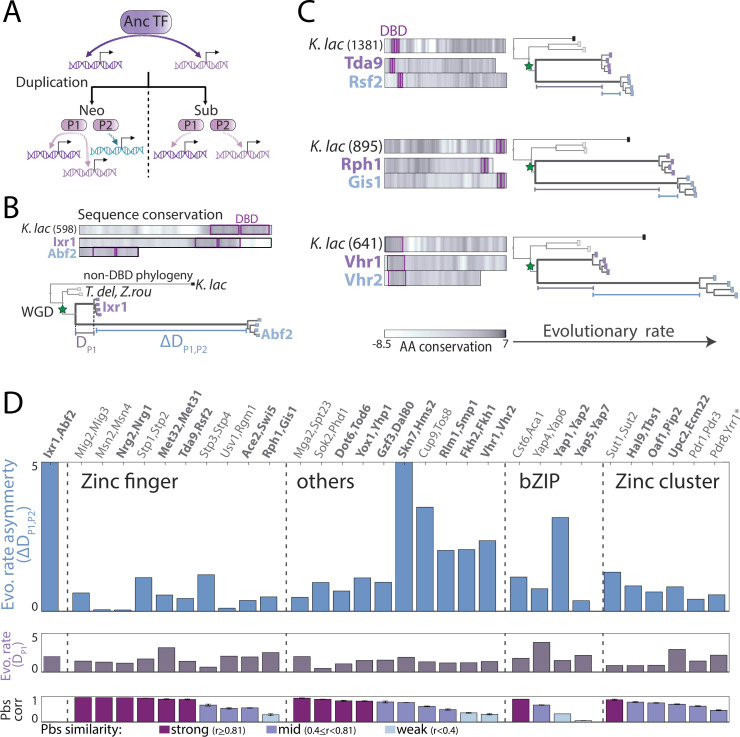
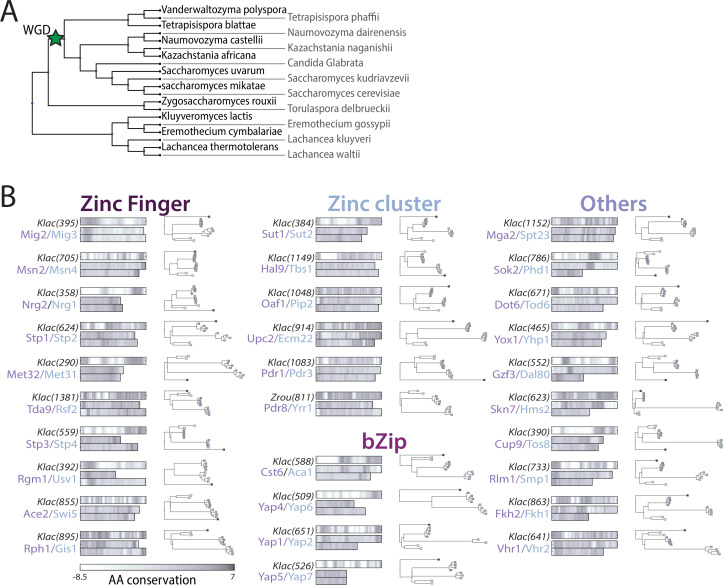
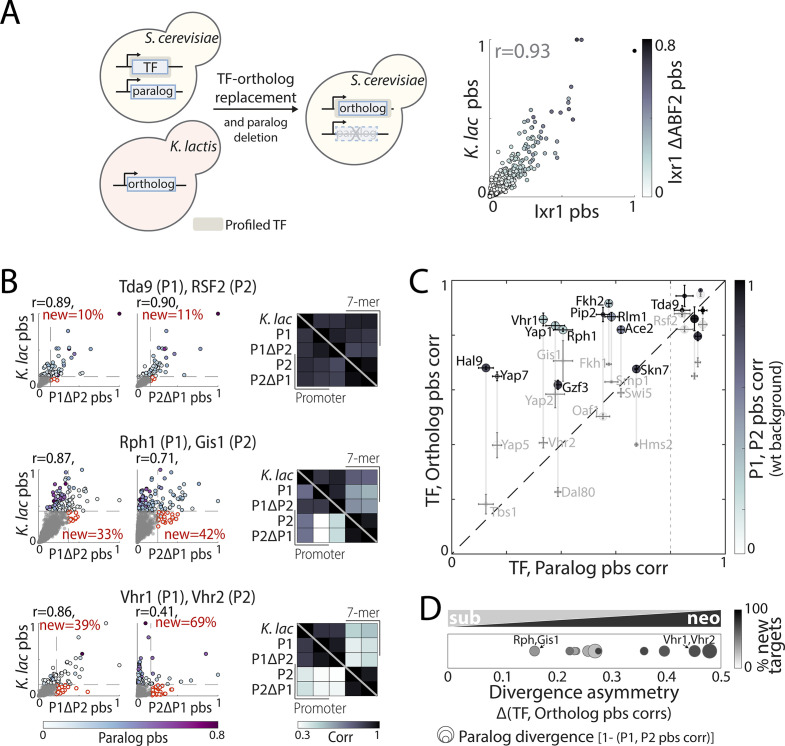
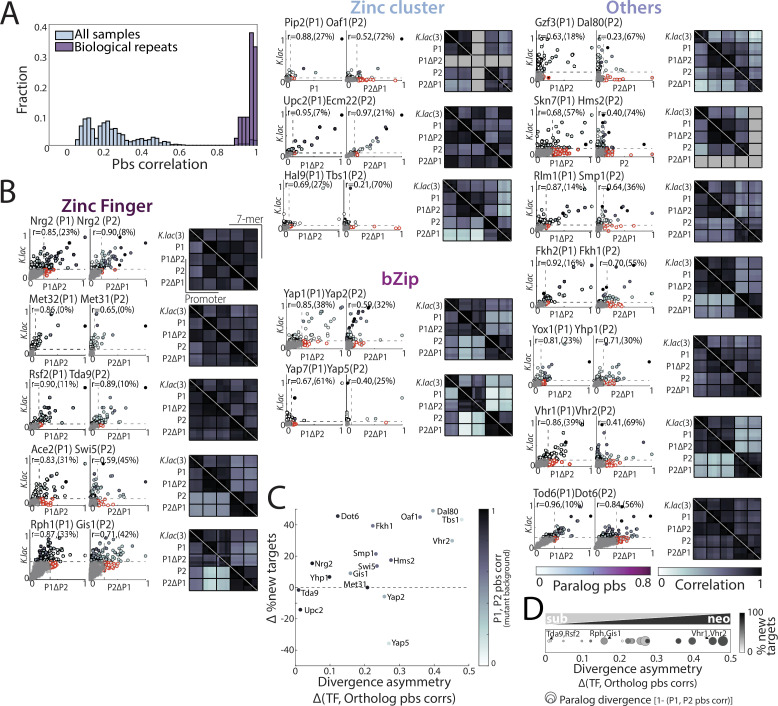
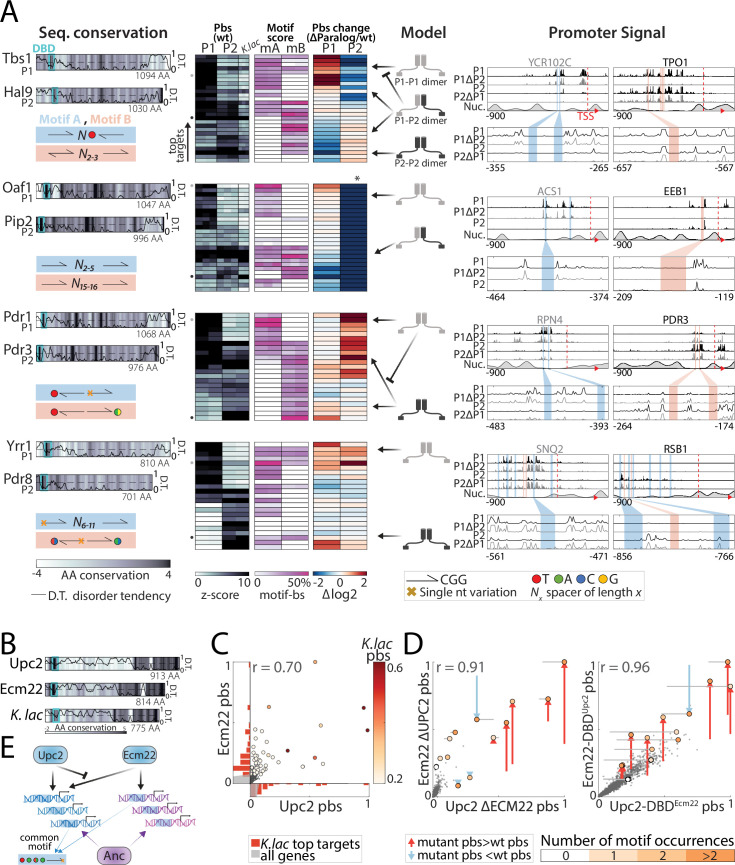
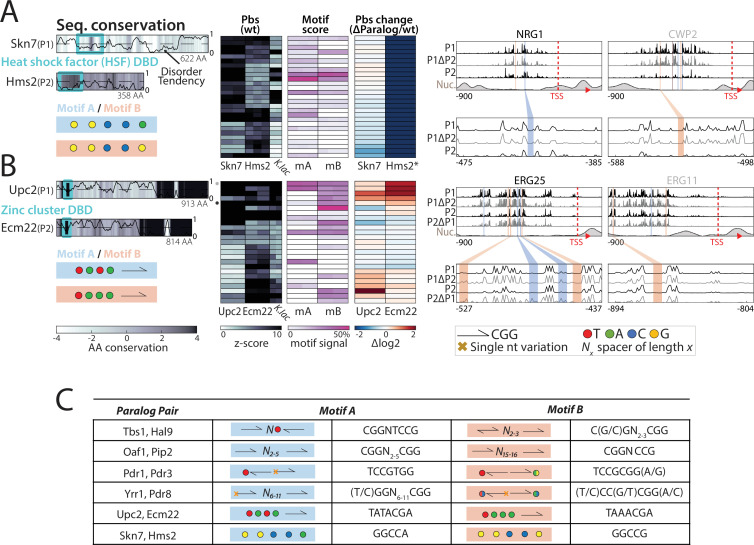
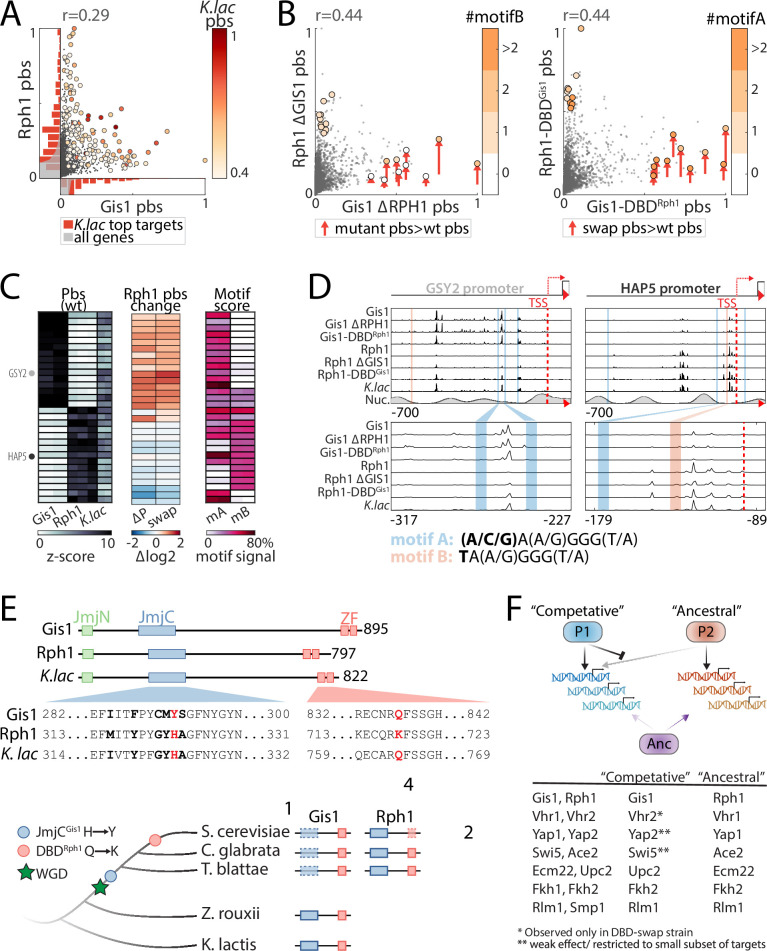
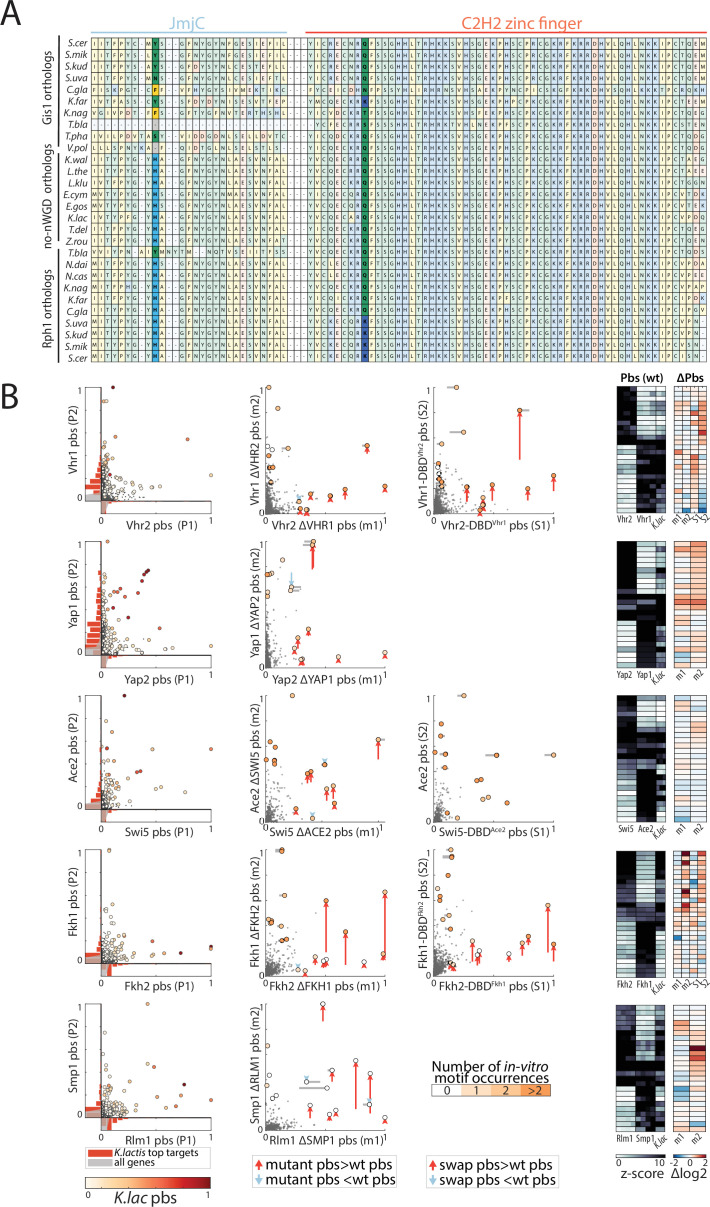
Throughout evolution, new transcription factors (TFs) emerge by gene duplication, promoting growth and rewiring of transcriptional networks. How TF duplicates diverge was studied in a few cases only. To provide a genome-scale view, we considered the set of budding yeast TFs classified as whole-genome duplication (WGD)-retained paralogs (~35% of all specific TFs). Using high-resolution profiling, we find that ~60% of paralogs evolved differential binding preferences. We show that this divergence results primarily from variations outside the DNA-binding domains (DBDs), while DBD preferences remain largely conserved. Analysis of non-WGD orthologs revealed uneven splitting of ancestral preferences between duplicates, and the preferential acquiring of new targets by the least conserved paralog (biased neo/sub-functionalization). Interactions between paralogs were rare, and, when present, occurred through weak competition for DNA-binding or dependency between dimer-forming paralogs. We discuss the implications of our findings for the evolutionary design of transcriptional networks.
Keywords: DNA binding; S. cerevisiae; evolutionary biology; functional divergence; gene regulation; genetics; genomics; paralogs; transcription factors; whole genome duplication.
© 2022, Gera et al.
Conflict of interest statement
TG, FJ, RM No competing interests declared, NB Senior editor, eLife
Figures

References
-
- Anand R, Memisoglu G, Haber J. Cas9-mediated gene editing in Saccharomyces cerevisiae. Protocol Exchange. 2017;1:e21. doi: 10.1038/protex.2017.021a. - DOI
-
- Baker Brachmann C, Davies A, Cost GJ, Caputo E, Li J, Hieter P, Boeke JD. Designer deletion strains derived from Saccharomyces cerevisiae S288C: A useful set of strains and plasmids for PCR-mediated gene disruption and other applications. Yeast (Chichester, England) 1998;14:115–132. doi: 10.1002/(SICI)1097-0061(19980130)14:2<115::AID-YEA204>3.0.CO;2-2. - DOI - PubMed
-
- Berger MF, Badis G, Gehrke AR, Talukder S, Philippakis AA, Peña-Castillo L, Alleyne TM, Mnaimneh S, Botvinnik OB, Chan ET, Khalid F, Zhang W, Newburger D, Jaeger SA, Morris QD, Bulyk ML, Hughes TR. Variation in homeodomain DNA binding revealed by high-resolution analysis of sequence preferences. Cell. 2008;133:1266–1276. doi: 10.1016/j.cell.2008.05.024. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Dryad/10.5061/dryad.xgxd254j6
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
