

Whole Genome Sequencing, Focused Assays and Functional Studies Increasing Understanding in Cryptic Inherited Retinal Dystrophies
- PMID: 35409265
- PMCID: PMC8999823
- DOI: 10.3390/ijms23073905
Whole Genome Sequencing, Focused Assays and Functional Studies Increasing Understanding in Cryptic Inherited Retinal Dystrophies
Abstract
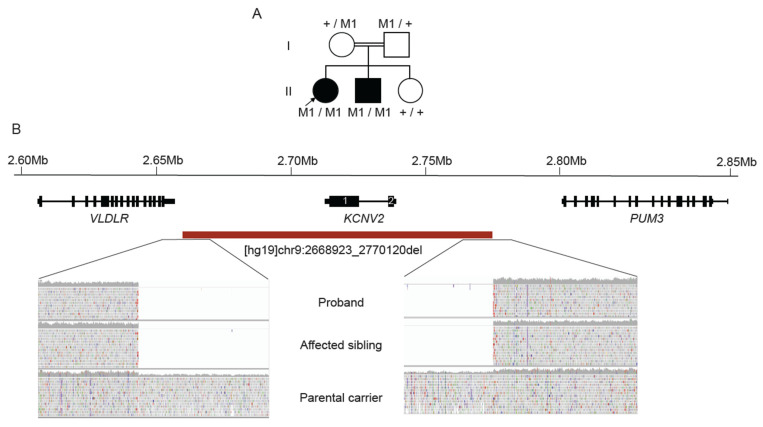
The inherited retinal dystrophies (IRDs) are a clinically and genetically complex group of disorders primarily affecting the rod and cone photoreceptors or other retinal neuronal layers, with emerging therapies heralding the need for accurate molecular diagnosis. Targeted capture and panel-based strategies examining the partial or full exome deliver molecular diagnoses in many IRD families tested. However, approximately one in three families remain unsolved and unable to obtain personalised recurrence risk or access to new clinical trials or therapy. In this study, we investigated whole genome sequencing (WGS), focused assays and functional studies to assist with unsolved IRD cases and facilitate integration of these approaches to a broad molecular diagnostic clinical service. The WGS approach identified variants not covered or underinvestigated by targeted capture panel-based clinical testing strategies in six families. This included structural variants, with notable benefit of the WGS approach in repetitive regions demonstrated by a family with a hybrid gene and hemizygous missense variant involving the opsin genes, OPN1LW and OPN1MW. There was also benefit in investigation of the repetitive GC-rich ORF15 region of RPGR. Further molecular investigations were facilitated by focused assays in these regions. Deep intronic variants were identified in IQCB1 and ABCA4, with functional RNA based studies of the IQCB1 variant revealing activation of a cryptic splice acceptor site. While targeted capture panel-based methods are successful in achieving an efficient molecular diagnosis in a proportion of cases, this study highlights the additional benefit and clinical value that may be derived from WGS, focused assays and functional genomics in the highly heterogeneous IRDs.
Keywords: RNA analysis; gene panels; inherited retinal dystrophy; whole genome sequencing.
Conflict of interest statement
The authors declare that they have no conflicts of interest.
Figures




References
-
- Maquire A.M., Russell S., Wellman J.A., Chung D.C., Yu Z., Tillman A., Wittes J., Pappas J., Elci O., Marshall K.A., et al. Efficacy, Safety, and Durability of Voretigene Neparvovec-rzyl in RPE65 Mutations Associated Inherited Retinal Dystrophy. Ophthalmology. 2019;126:1273–1285. doi: 10.1016/j.ophtha.2019.06.017. - DOI - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
