

Exome sequencing in bipolar disorder identifies AKAP11 as a risk gene shared with schizophrenia
- PMID: 35410376
- PMCID: PMC9117467
- DOI: 10.1038/s41588-022-01034-x
Exome sequencing in bipolar disorder identifies AKAP11 as a risk gene shared with schizophrenia
Abstract
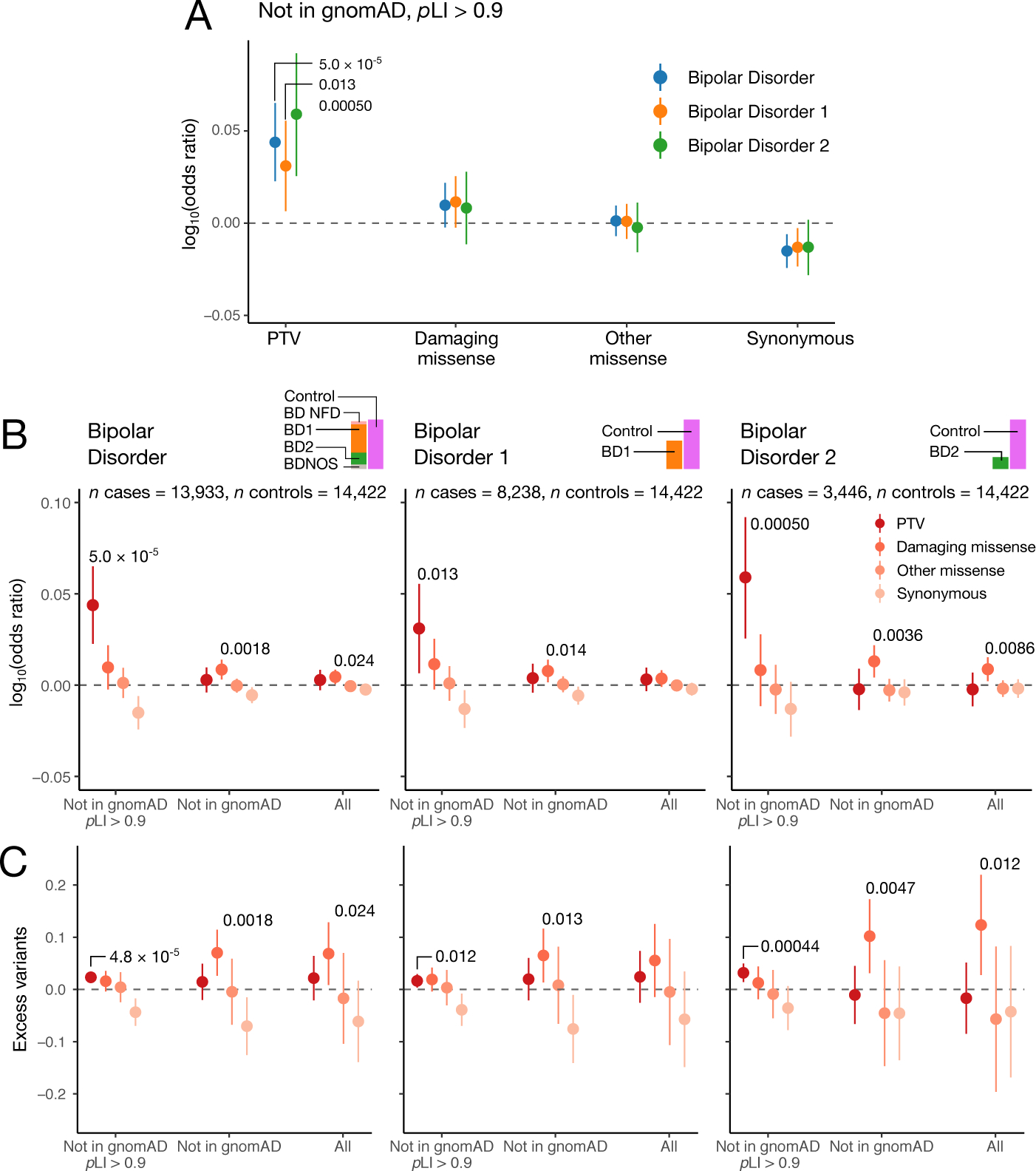
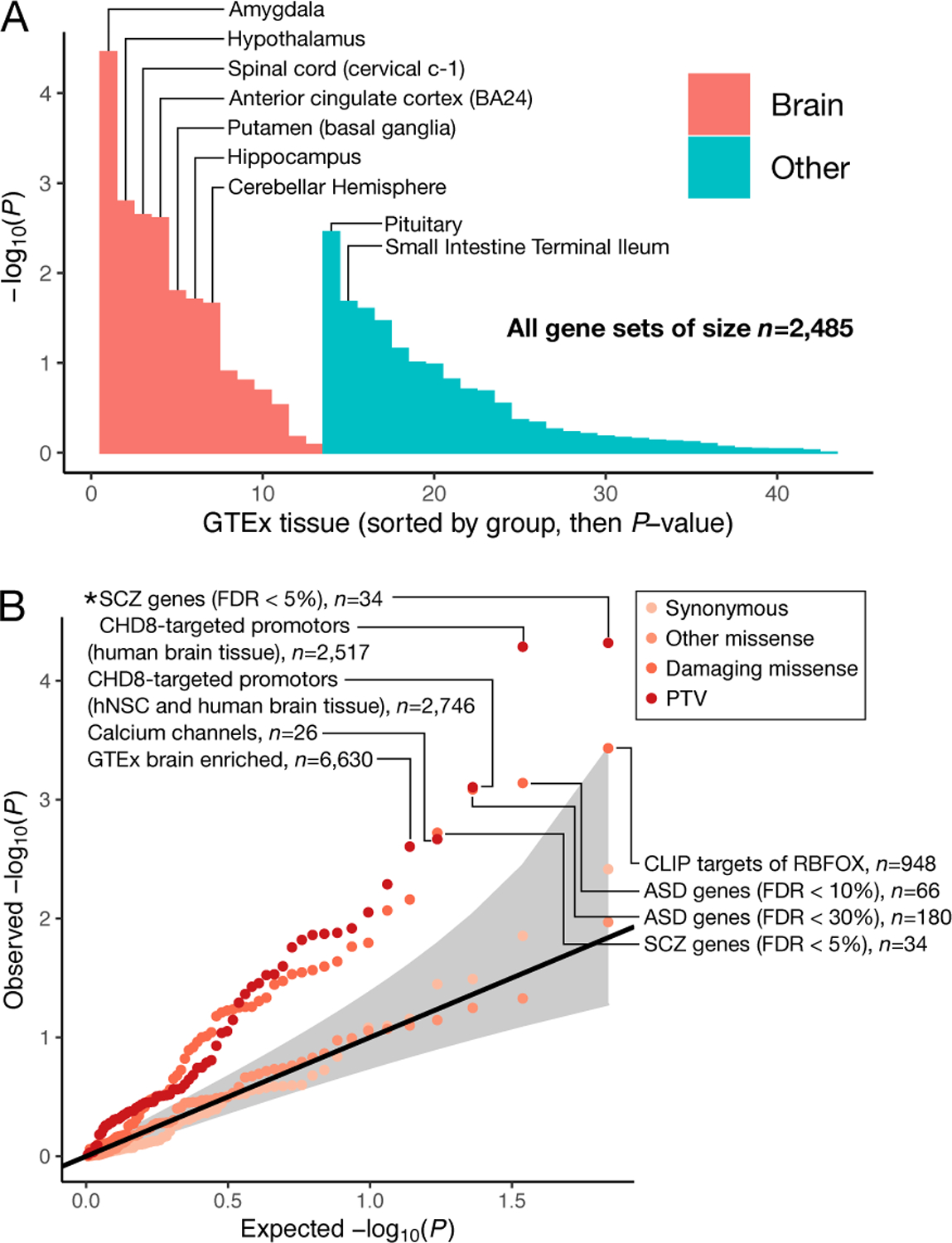
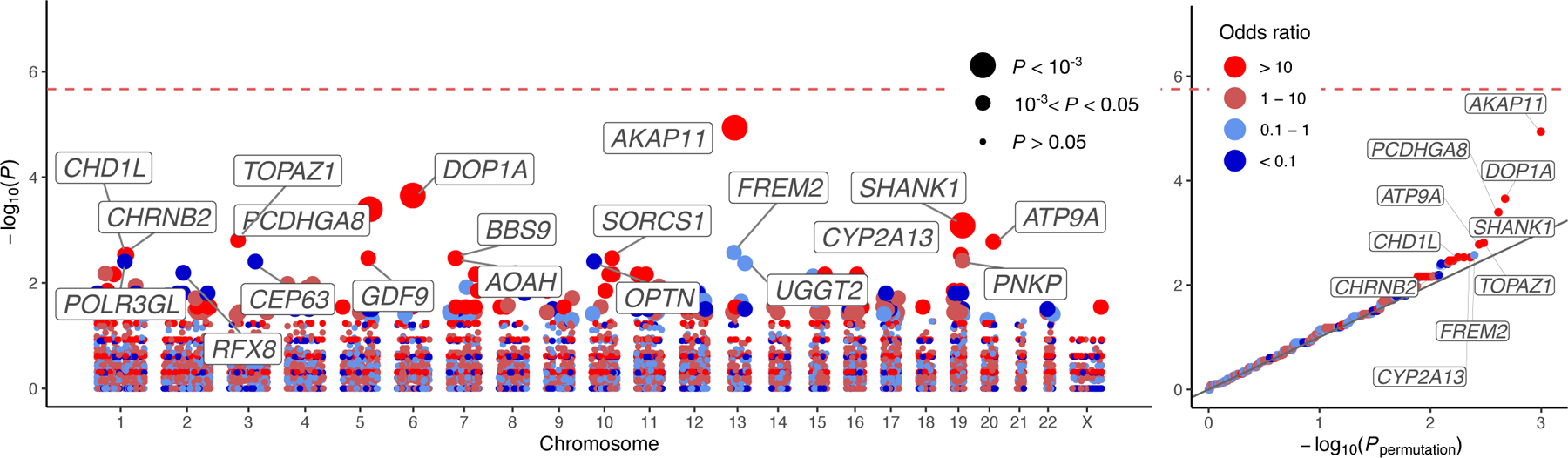
We report results from the Bipolar Exome (BipEx) collaboration analysis of whole-exome sequencing of 13,933 patients with bipolar disorder (BD) matched with 14,422 controls. We find an excess of ultra-rare protein-truncating variants (PTVs) in patients with BD among genes under strong evolutionary constraint in both major BD subtypes. We find enrichment of ultra-rare PTVs within genes implicated from a recent schizophrenia exome meta-analysis (SCHEMA; 24,248 cases and 97,322 controls) and among binding targets of CHD8. Genes implicated from genome-wide association studies (GWASs) of BD, however, are not significantly enriched for ultra-rare PTVs. Combining gene-level results with SCHEMA, AKAP11 emerges as a definitive risk gene (odds ratio (OR) = 7.06, P = 2.83 × 10-9). At the protein level, AKAP-11 interacts with GSK3B, the hypothesized target of lithium, a primary treatment for BD. Our results lend support to BD's polygenicity, demonstrating a role for rare coding variation as a significant risk factor in BD etiology.
© 2022. The Author(s), under exclusive licence to Springer Nature America, Inc.
Figures
References
-
- Ferrari AJ et al. The prevalence and burden of bipolar disorder: findings from the Global Burden of Disease Study 2013. Bipolar Disord. 18, 440–450 (2016). - PubMed
-
- Polderman TJC et al. Meta-analysis of the heritability of human traits based on fifty years of twin studies. Nat. Genet. 47, 702–709 (2015). - PubMed
Methods references
Publication types
MeSH terms
Substances
Grants and funding
- R01 MH110437/MH/NIMH NIH HHS/United States
- MR/P005748/1/MRC_/Medical Research Council/United Kingdom
- G1000708/MRC_/Medical Research Council/United Kingdom
- R01 CA194393/CA/NCI NIH HHS/United States
- MR/L010305/1/MRC_/Medical Research Council/United Kingdom
- R01 MH085542/MH/NIMH NIH HHS/United States
- R01 MH090553/MH/NIMH NIH HHS/United States
- RC2 AG036607/AG/NIA NIH HHS/United States
- R01 MH095034/MH/NIMH NIH HHS/United States
- R01 MH085543/MH/NIMH NIH HHS/United States
- R37 MH107649/MH/NIMH NIH HHS/United States
- U01 MH105578/MH/NIMH NIH HHS/United States
- R01 MH084676/MH/NIMH NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
