

Management challenges and therapeutic advances in congenital adrenal hyperplasia
- PMID: 35411073
- PMCID: PMC8999997
- DOI: 10.1038/s41574-022-00655-w
Management challenges and therapeutic advances in congenital adrenal hyperplasia
Abstract
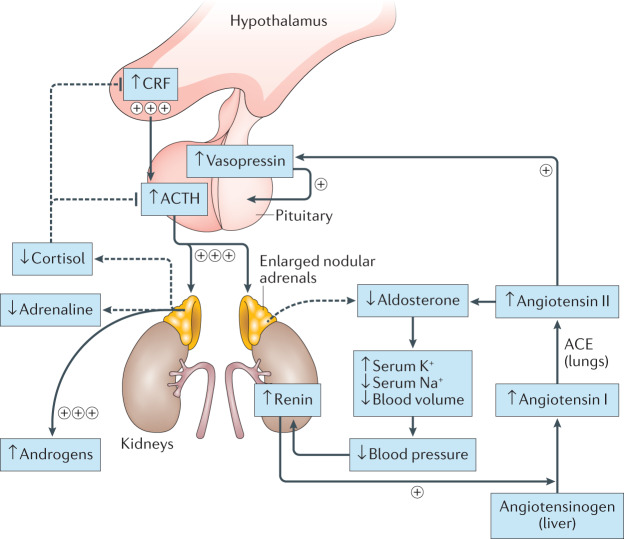
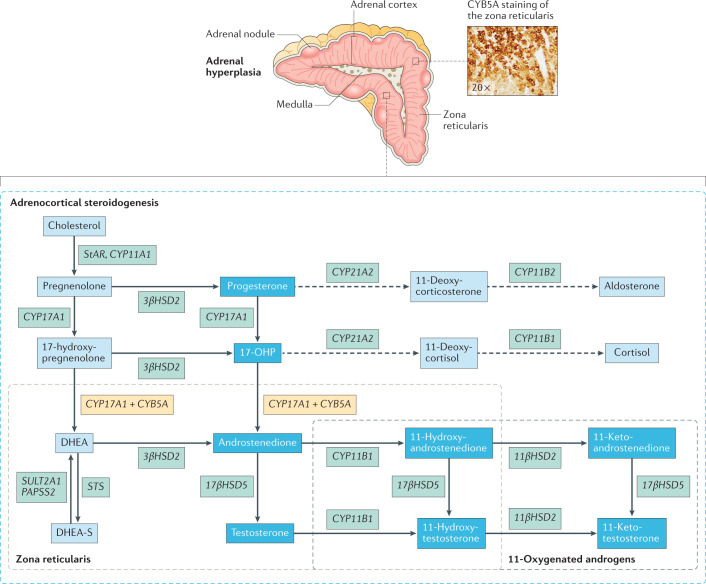
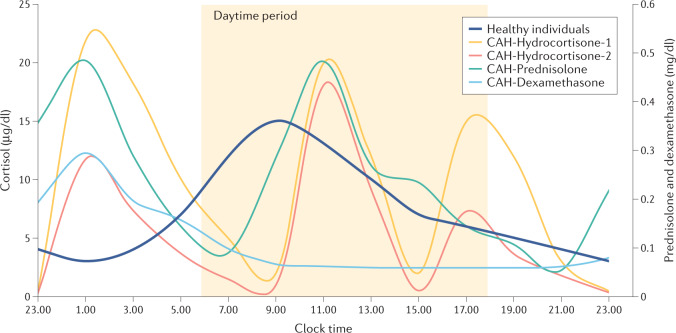
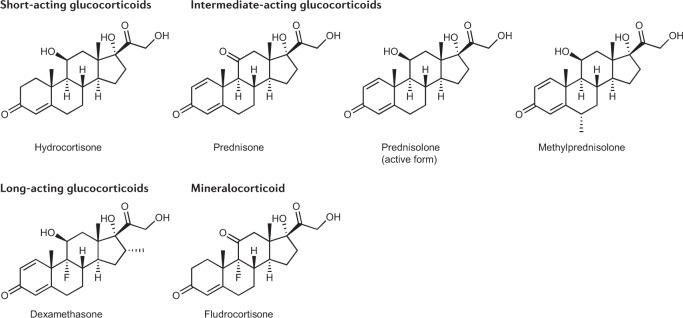
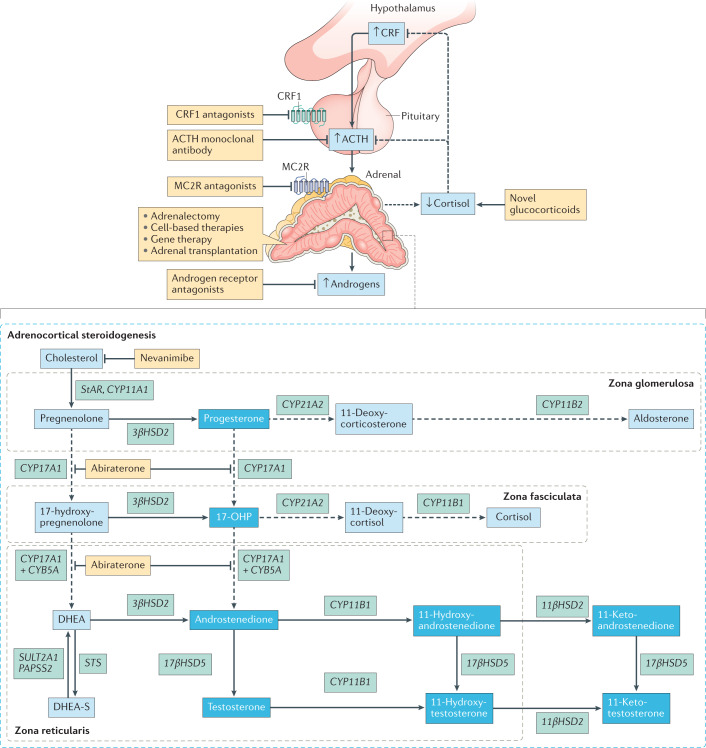
Treatment for congenital adrenal hyperplasia (CAH) was introduced in the 1950s following the discovery of the structure and function of adrenocortical hormones. Although major advances in molecular biology have delineated steroidogenic mechanisms and the genetics of CAH, management and treatment of this condition continue to present challenges. Management is complicated by a combination of comorbidities that arise from disease-related hormonal derangements and treatment-related adverse effects. The clinical outcomes of CAH can include life-threatening adrenal crises, altered growth and early puberty, and adverse effects on metabolic, cardiovascular, bone and reproductive health. Standard-of-care glucocorticoid formulations fall short of replicating the circadian rhythm of cortisol and controlling efficient adrenocorticotrophic hormone-driven adrenal androgen production. Adrenal-derived 11-oxygenated androgens have emerged as potential new biomarkers for CAH, as traditional biomarkers are subject to variability and are not adrenal-specific, contributing to management challenges. Multiple alternative treatment approaches are being developed with the aim of tailoring therapy for improved patient outcomes. This Review focuses on challenges and advances in the management and treatment of CAH due to 21-hydroxylase deficiency, the most common type of CAH. Furthermore, we examine new therapeutic developments, including treatments designed to replace cortisol in a physiological manner and adjunct agents intended to control excess androgens and thereby enable reductions in glucocorticoid doses.
© 2022. This is a U.S. government work and not under copyright protection in the U.S.; foreign copyright protection may apply.
Conflict of interest statement
D.P.M. received research funds from Diurnal Limited through the National Institutes of Health Cooperative Research and Development Agreement. During the writing of this manuscript, A.M. took up employment at AstraZeneca and is currently employed there.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
