

A molecular complex of Cav1.2/CaMKK2/CaMK1a in caveolae is responsible for vascular remodeling via excitation-transcription coupling
- PMID: 35412911
- PMCID: PMC9169798
- DOI: 10.1073/pnas.2117435119
A molecular complex of Cav1.2/CaMKK2/CaMK1a in caveolae is responsible for vascular remodeling via excitation-transcription coupling
Abstract
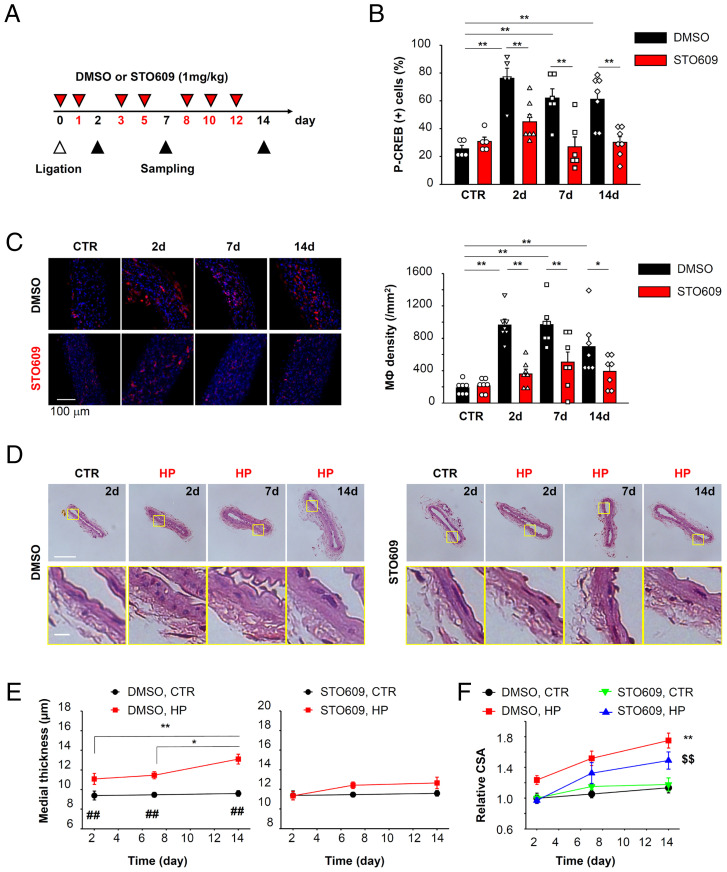
Elevation of intracellular Ca2+ concentration ([Ca2+]i) activates Ca2+/calmodulin-dependent kinases (CaMK) and promotes gene transcription. This signaling pathway is referred to as excitation–transcription (E-T) coupling. Although vascular myocytes can exhibit E-T coupling, the molecular mechanisms and physiological/pathological roles are unknown. Multiscale analysis spanning from single molecules to whole organisms has revealed essential steps in mouse vascular myocyte E-T coupling. Upon a depolarizing stimulus, Ca2+ influx through Cav1.2 voltage-dependent Ca2+ channels activates CaMKK2 and CaMK1a, resulting in intranuclear CREB phosphorylation. Within caveolae, the formation of a molecular complex of Cav1.2/CaMKK2/CaMK1a is promoted in vascular myocytes. Live imaging using a genetically encoded Ca2+ indicator revealed direct activation of CaMKK2 by Ca2+ influx through Cav1.2 localized to caveolae. CaMK1a is phosphorylated by CaMKK2 at caveolae and translocated to the nucleus upon membrane depolarization. In addition, sustained depolarization of a mesenteric artery preparation induced genes related to chemotaxis, leukocyte adhesion, and inflammation, and these changes were reversed by inhibitors of Cav1.2, CaMKK2, and CaMK, or disruption of caveolae. In the context of pathophysiology, when the mesenteric artery was loaded by high pressure in vivo, we observed CREB phosphorylation in myocytes, macrophage accumulation at adventitia, and an increase in thickness and cross-sectional area of the tunica media. These changes were reduced in caveolin1-knockout mice or in mice treated with the CaMKK2 inhibitor STO609. In summary, E-T coupling depends on Cav1.2/CaMKK2/CaMK1a localized to caveolae, and this complex converts [Ca2+]i changes into gene transcription. This ultimately leads to macrophage accumulation and media remodeling for adaptation to increased circumferential stretch.
Keywords: caveolin-1; excitation–transcription coupling; vascular remodeling; vascular smooth muscle cell; voltage-dependent Ca2+ channel.
Conflict of interest statement
The authors declare no competing interest.
Figures








Comment in
-
Vasculature remodeling by pressure, caveolae, calcium, and kinases.Proc Natl Acad Sci U S A. 2022 May 24;119(21):e2204968119. doi: 10.1073/pnas.2204968119. Epub 2022 May 18. Proc Natl Acad Sci U S A. 2022. PMID: 35584115 Free PMC article. No abstract available.
Similar articles
-
Local Ca2+ Signals within Caveolae Cause Nuclear Translocation of CaMK1α in Mouse Vascular Smooth Muscle Cells.Biol Pharm Bull. 2022;45(9):1354-1363. doi: 10.1248/bpb.b22-00319. Biol Pharm Bull. 2022. PMID: 36047205
-
Caveolin-1 facilitates the direct coupling between large conductance Ca2+-activated K+ (BKCa) and Cav1.2 Ca2+ channels and their clustering to regulate membrane excitability in vascular myocytes.J Biol Chem. 2013 Dec 20;288(51):36750-61. doi: 10.1074/jbc.M113.511485. Epub 2013 Nov 7. J Biol Chem. 2013. PMID: 24202214 Free PMC article.
-
PKCζ-NADPH Oxidase-PKCα Dependent Kv1.5 Phosphorylation by Endothelin-1 Modulates Nav1.5-NCX1-Cav1.2 Axis in Stimulating Ca2+ Level in Caveolae of Pulmonary Artery Smooth Muscle Cells.Cell Biochem Biophys. 2021 Mar;79(1):57-71. doi: 10.1007/s12013-020-00954-x. Epub 2020 Oct 23. Cell Biochem Biophys. 2021. PMID: 33095400
-
CaV1.2 sparklets in heart and vascular smooth muscle.J Mol Cell Cardiol. 2013 May;58:67-76. doi: 10.1016/j.yjmcc.2012.11.018. Epub 2012 Dec 6. J Mol Cell Cardiol. 2013. PMID: 23220157 Free PMC article. Review.
-
Excitation-transcription coupling in arterial smooth muscle.Circ Res. 2006 Apr 14;98(7):868-78. doi: 10.1161/01.RES.0000216596.73005.3c. Circ Res. 2006. PMID: 16614312 Review.
Cited by
-
A sonoelectric niche for noninvasive intervertebral disc regeneration via targeted cell cycle modulation.Sci Adv. 2025 Aug 8;11(32):eadu6860. doi: 10.1126/sciadv.adu6860. Epub 2025 Aug 8. Sci Adv. 2025. PMID: 40779637 Free PMC article.
-
Molecular Mechanisms Underlying Ca2+/Calmodulin-Dependent Protein Kinase Kinase Signal Transduction.Int J Mol Sci. 2022 Sep 20;23(19):11025. doi: 10.3390/ijms231911025. Int J Mol Sci. 2022. PMID: 36232320 Free PMC article. Review.
-
The role of CaMKK2 in Golgi-associated vesicle trafficking.Biochem Soc Trans. 2023 Feb 27;51(1):331-342. doi: 10.1042/BST20220833. Biochem Soc Trans. 2023. PMID: 36815702 Free PMC article. Review.
-
Vasculature remodeling by pressure, caveolae, calcium, and kinases.Proc Natl Acad Sci U S A. 2022 May 24;119(21):e2204968119. doi: 10.1073/pnas.2204968119. Epub 2022 May 18. Proc Natl Acad Sci U S A. 2022. PMID: 35584115 Free PMC article. No abstract available.
-
Ca2+ microdomain-based excitation-transcription coupling in cardiac myocytes and vascular smooth muscle cells.Inflamm Regen. 2025 Jun 23;45(1):19. doi: 10.1186/s41232-025-00384-3. Inflamm Regen. 2025. PMID: 40551210 Free PMC article. Review.
References
-
- Berridge M. J., The inositol trisphosphate/calcium signaling pathway in health and disease. Physiol. Rev. 96, 1261–1296 (2016). - PubMed
-
- Bers D. M., Cardiac excitation-contraction coupling. Nature 415, 198–205 (2002). - PubMed
-
- Jaggar J. H., Porter V. A., Lederer W. J., Nelson M. T., Calcium sparks in smooth muscle. Am. J. Physiol. Cell Physiol. 278, C235–C256 (2000). - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
