

Population-scale long-read sequencing uncovers transposable elements associated with gene expression variation and adaptive signatures in Drosophila
- PMID: 35413957
- PMCID: PMC9005704
- DOI: 10.1038/s41467-022-29518-8
Population-scale long-read sequencing uncovers transposable elements associated with gene expression variation and adaptive signatures in Drosophila
Abstract
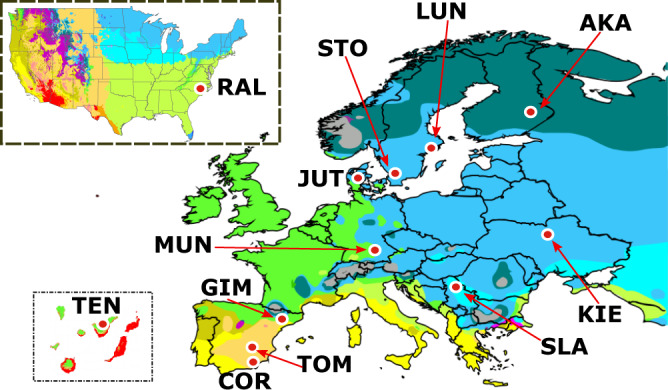
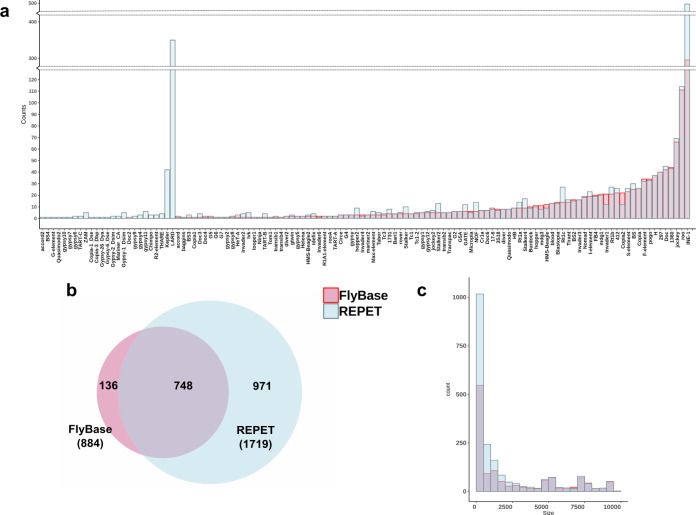
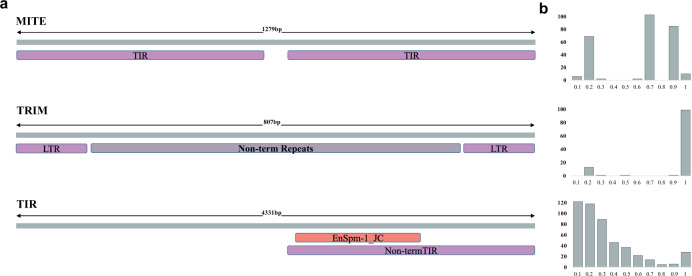
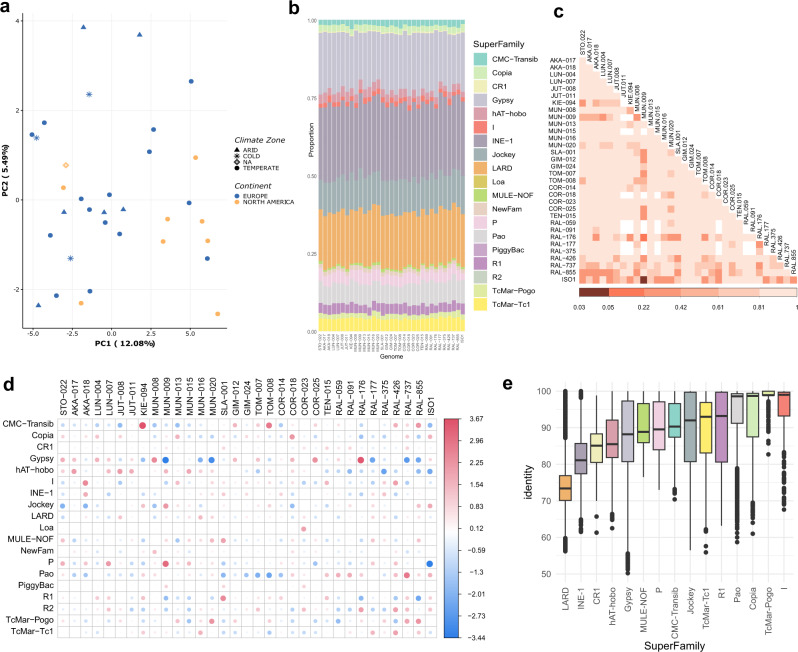
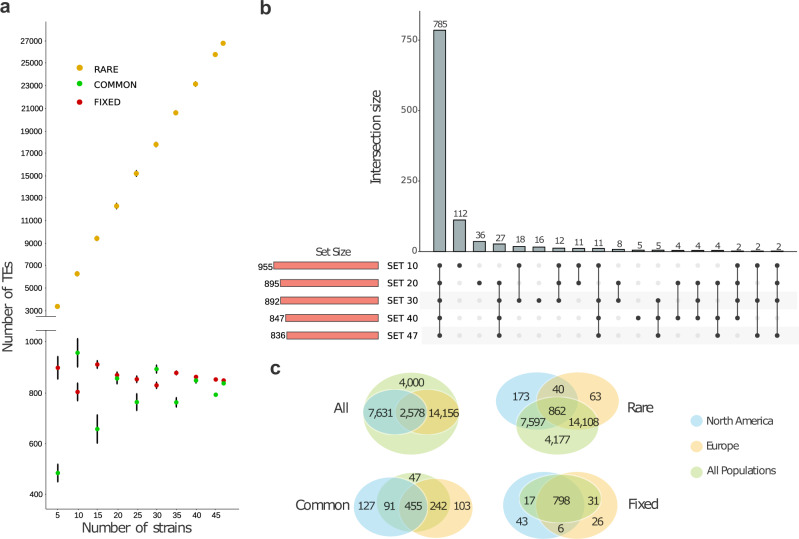
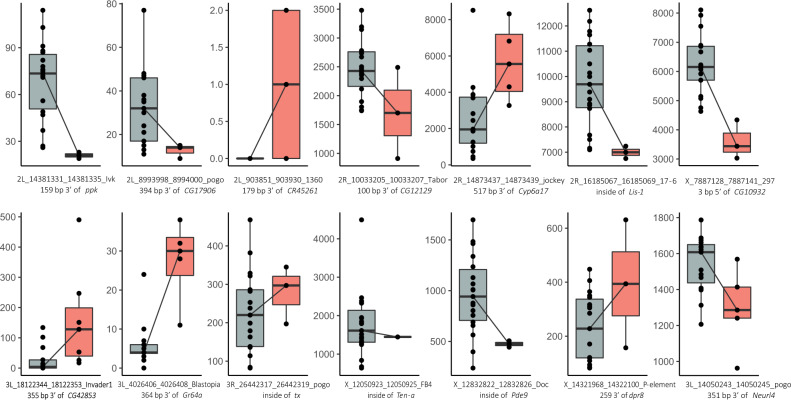
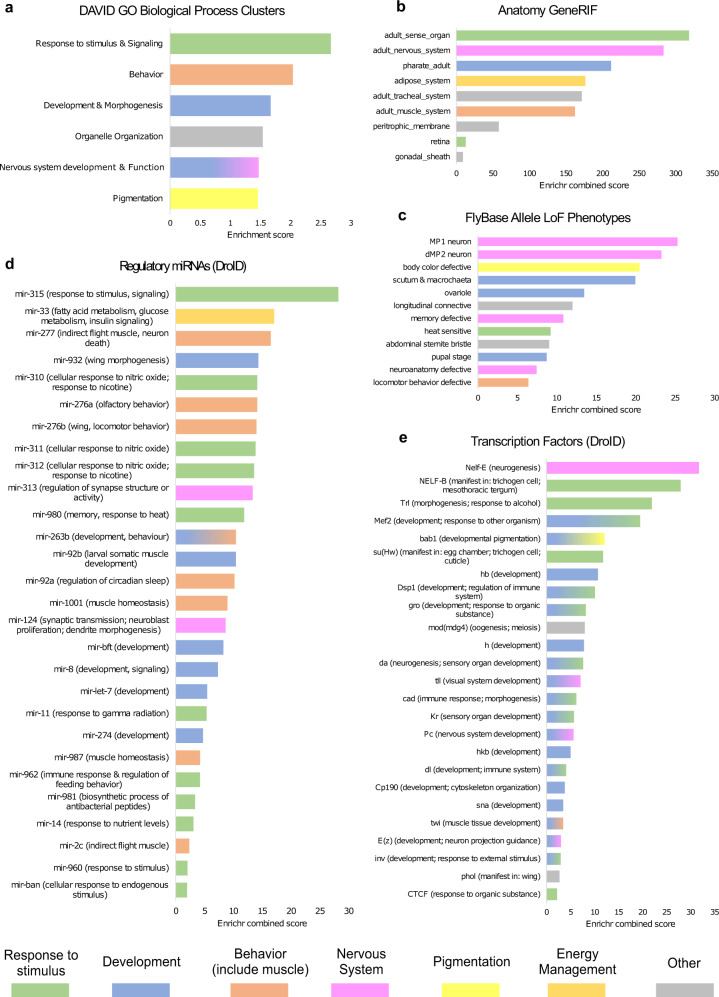
High quality reference genomes are crucial to understanding genome function, structure and evolution. The availability of reference genomes has allowed us to start inferring the role of genetic variation in biology, disease, and biodiversity conservation. However, analyses across organisms demonstrate that a single reference genome is not enough to capture the global genetic diversity present in populations. In this work, we generate 32 high-quality reference genomes for the well-known model species D. melanogaster and focus on the identification and analysis of transposable element variation as they are the most common type of structural variant. We show that integrating the genetic variation across natural populations from five climatic regions increases the number of detected insertions by 58%. Moreover, 26% to 57% of the insertions identified using long-reads were missed by short-reads methods. We also identify hundreds of transposable elements associated with gene expression variation and new TE variants likely to contribute to adaptive evolution in this species. Our results highlight the importance of incorporating the genetic variation present in natural populations to genomic studies, which is essential if we are to understand how genomes function and evolve.
© 2022. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- De Coster W, Van Broeckhoven C. Newest methods for detecting structural variations. Trends Biotechnol. 2019;37:973–982. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
