

A machine learning algorithm with subclonal sensitivity reveals widespread pan-cancer human leukocyte antigen loss of heterozygosity
- PMID: 35414054
- PMCID: PMC9005524
- DOI: 10.1038/s41467-022-29203-w
A machine learning algorithm with subclonal sensitivity reveals widespread pan-cancer human leukocyte antigen loss of heterozygosity
Abstract
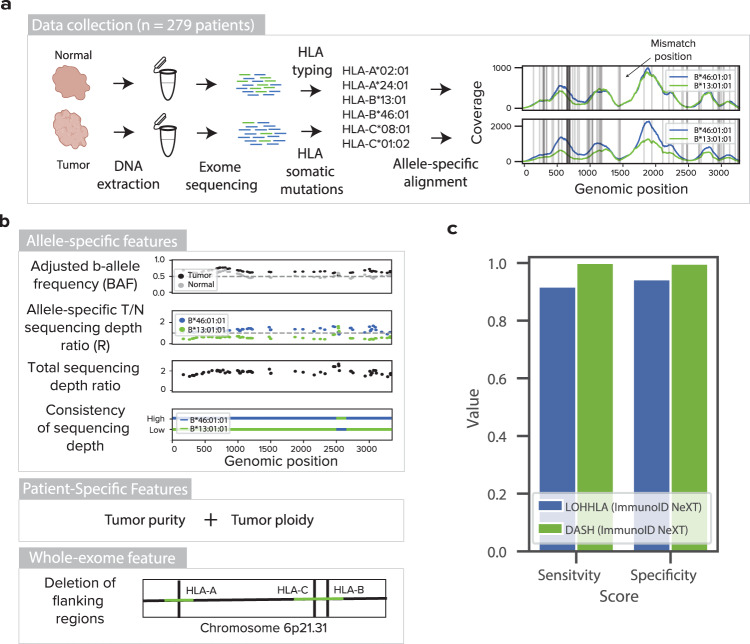
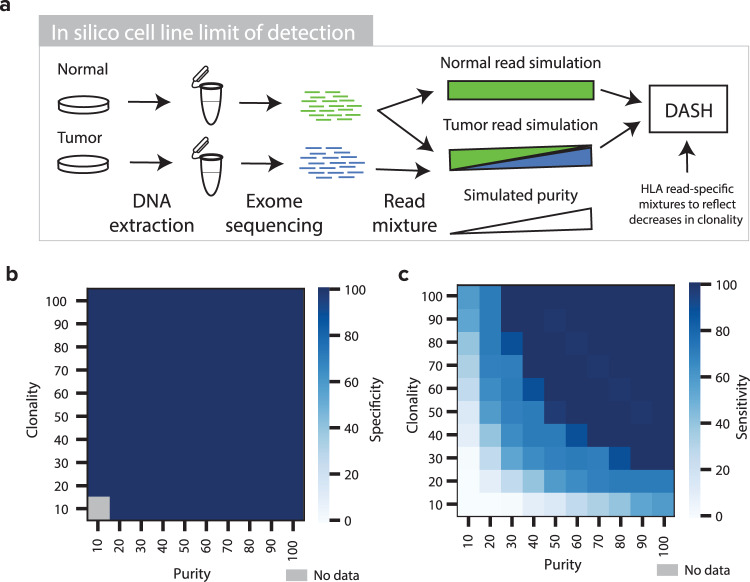
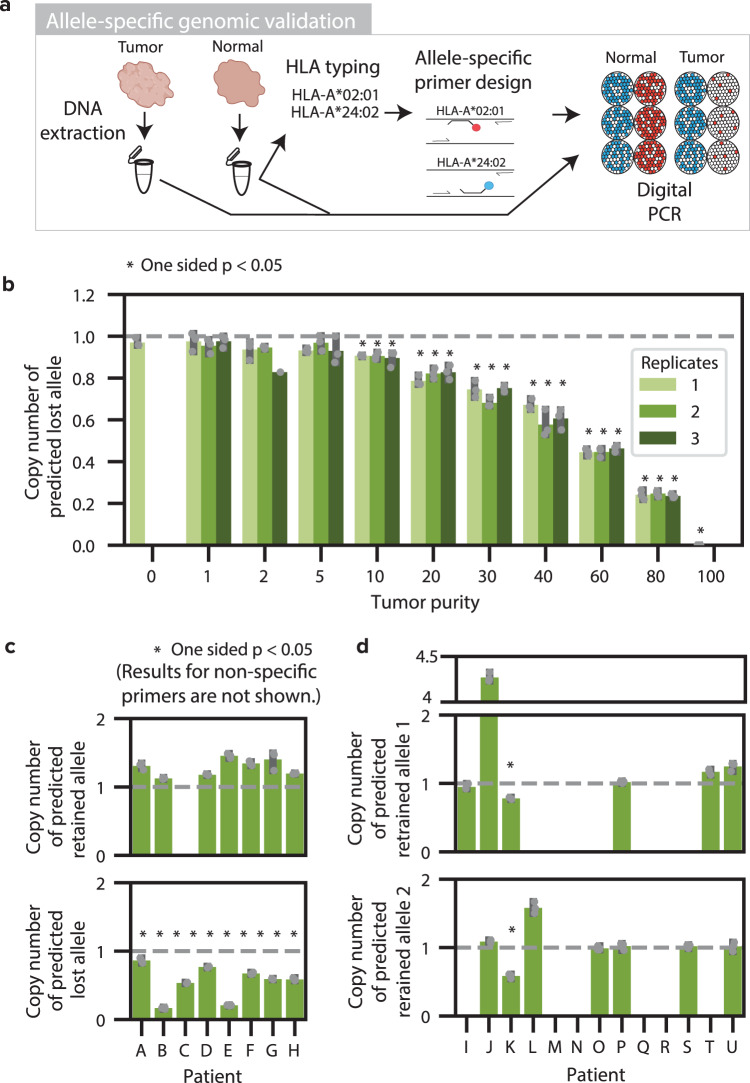
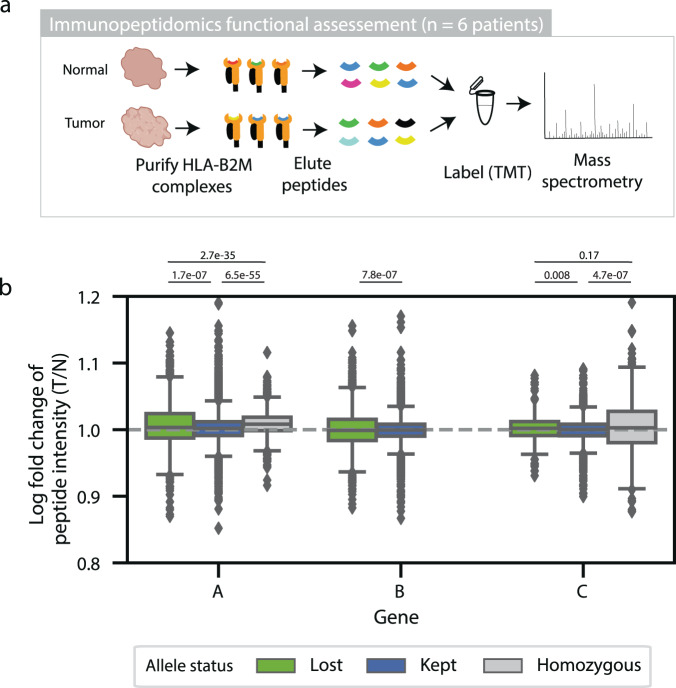
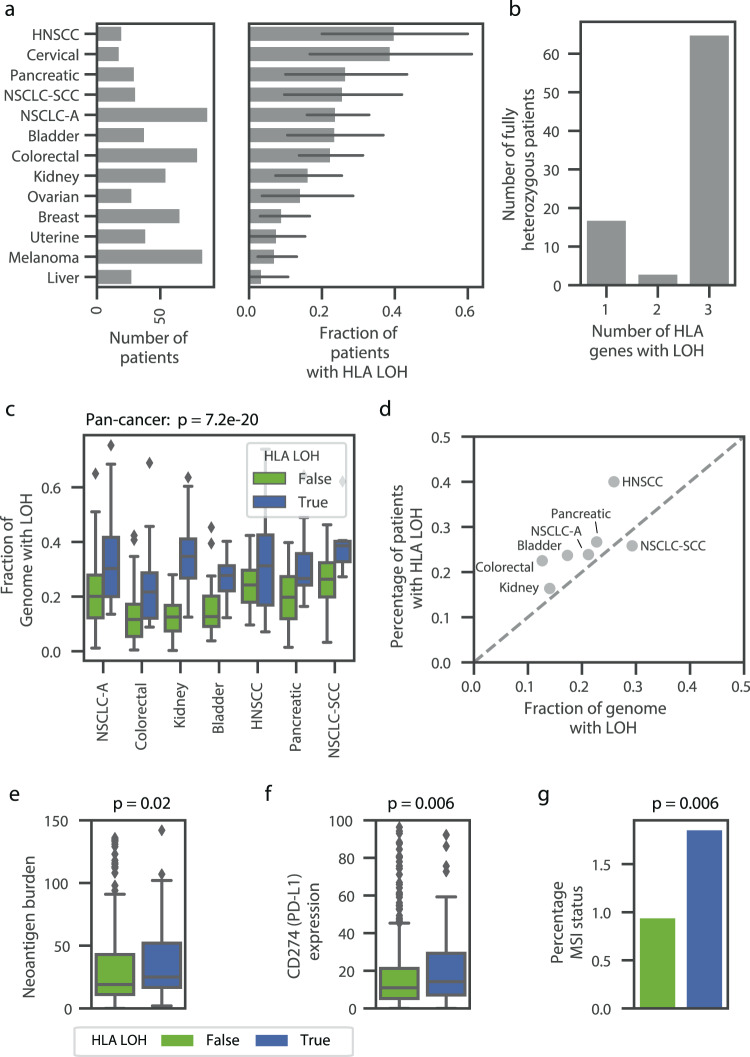
Human leukocyte antigen loss of heterozygosity (HLA LOH) allows cancer cells to escape immune recognition by deleting HLA alleles, causing the suppressed presentation of tumor neoantigens. Despite its importance in immunotherapy response, few methods exist to detect HLA LOH, and their accuracy is not well understood. Here, we develop DASH (Deletion of Allele-Specific HLAs), a machine learning-based algorithm to detect HLA LOH from paired tumor-normal sequencing data. With cell line mixtures, we demonstrate increased sensitivity compared to previously published tools. Moreover, our patient-specific digital PCR validation approach provides a sensitive, robust orthogonal approach that could be used for clinical validation. Using DASH on 610 patients across 15 tumor types, we find that 18% of patients have HLA LOH. Moreover, we show inflated HLA LOH rates compared to genome-wide LOH and correlations between CD274 (encodes PD-L1) expression and microsatellite instability status, suggesting the HLA LOH is a key immune resistance strategy.
© 2022. The Author(s).
Conflict of interest statement
R.M.P., D.M., S.D., C.W.A., L.M., D.P.B., S.V.Z., E.L., G.B., J.W., R.O.C. and S.M.B. are full time employees of Personalis. M.P.S. co-founded and is a scientific advisor for Personalis.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
