

The interferon-stimulated gene RIPK1 regulates cancer cell intrinsic and extrinsic resistance to immune checkpoint blockade
- PMID: 35417675
- PMCID: PMC11289737
- DOI: 10.1016/j.immuni.2022.03.007
The interferon-stimulated gene RIPK1 regulates cancer cell intrinsic and extrinsic resistance to immune checkpoint blockade
Abstract
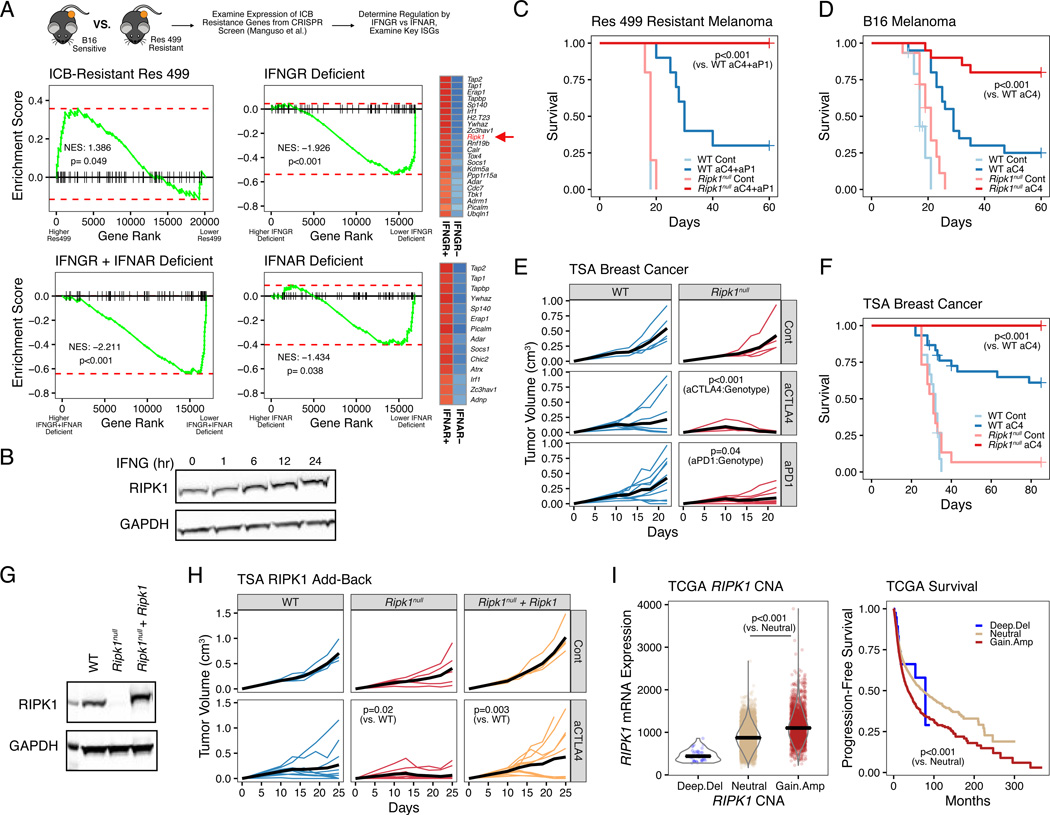
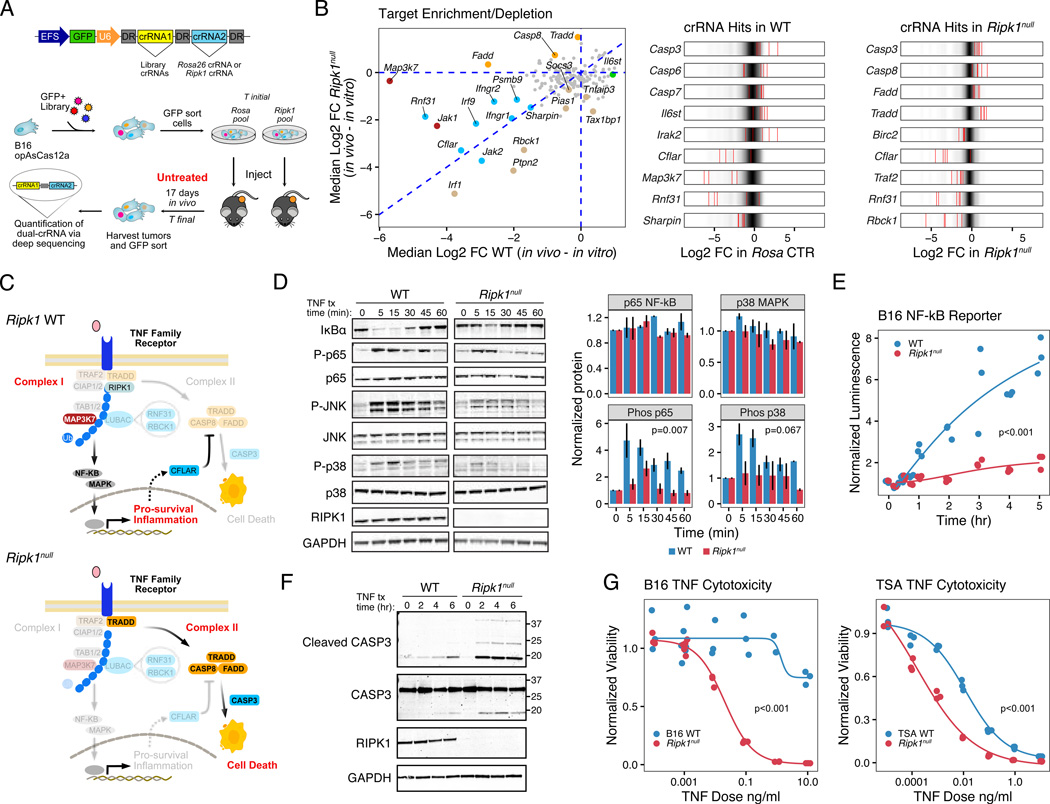
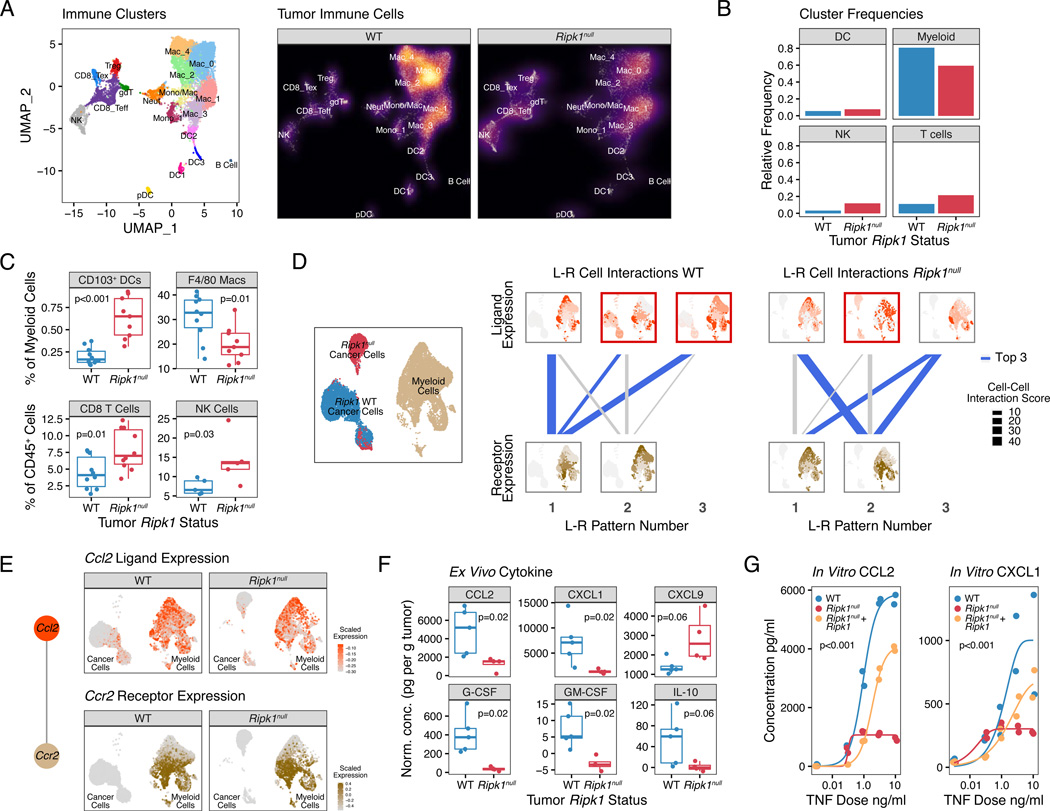
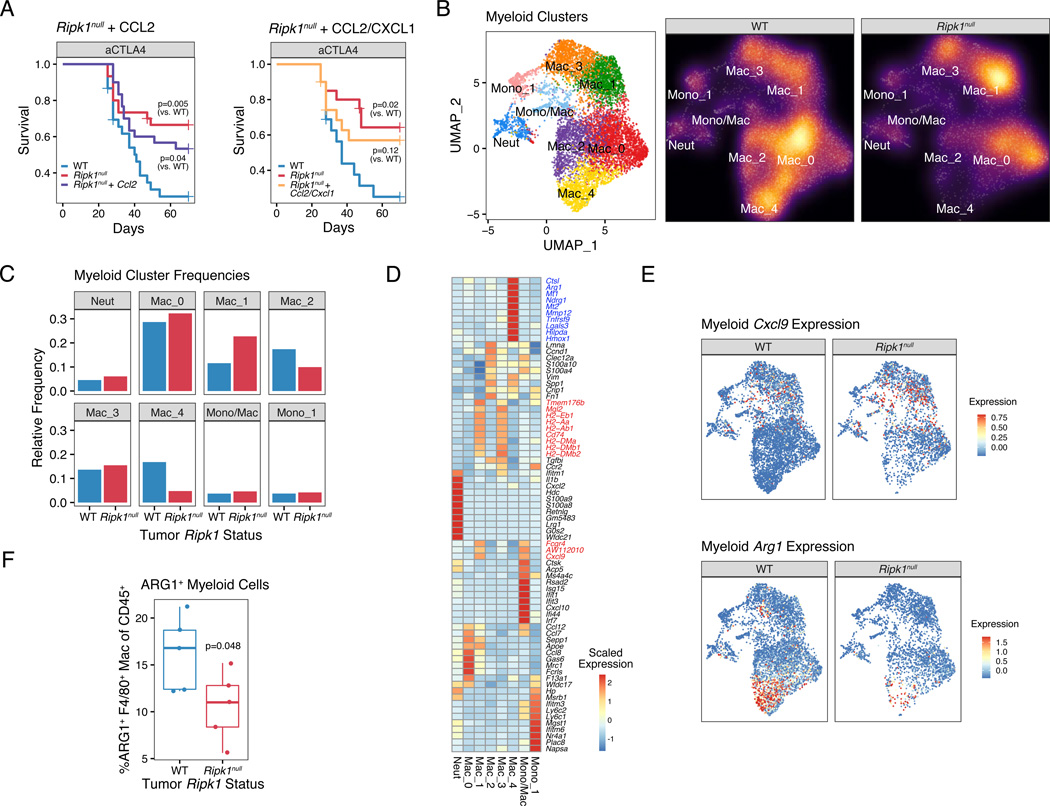
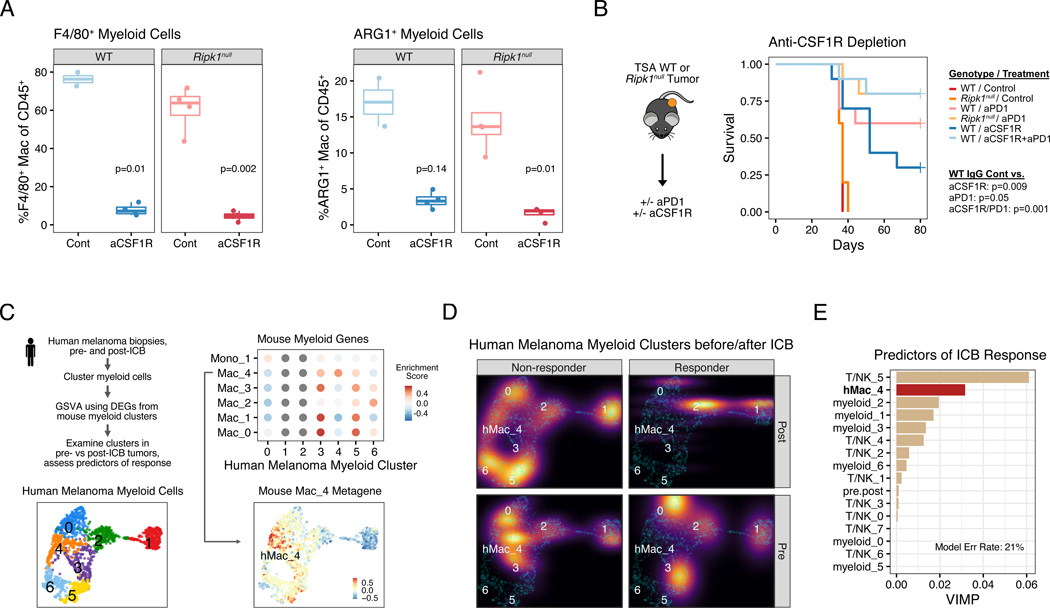
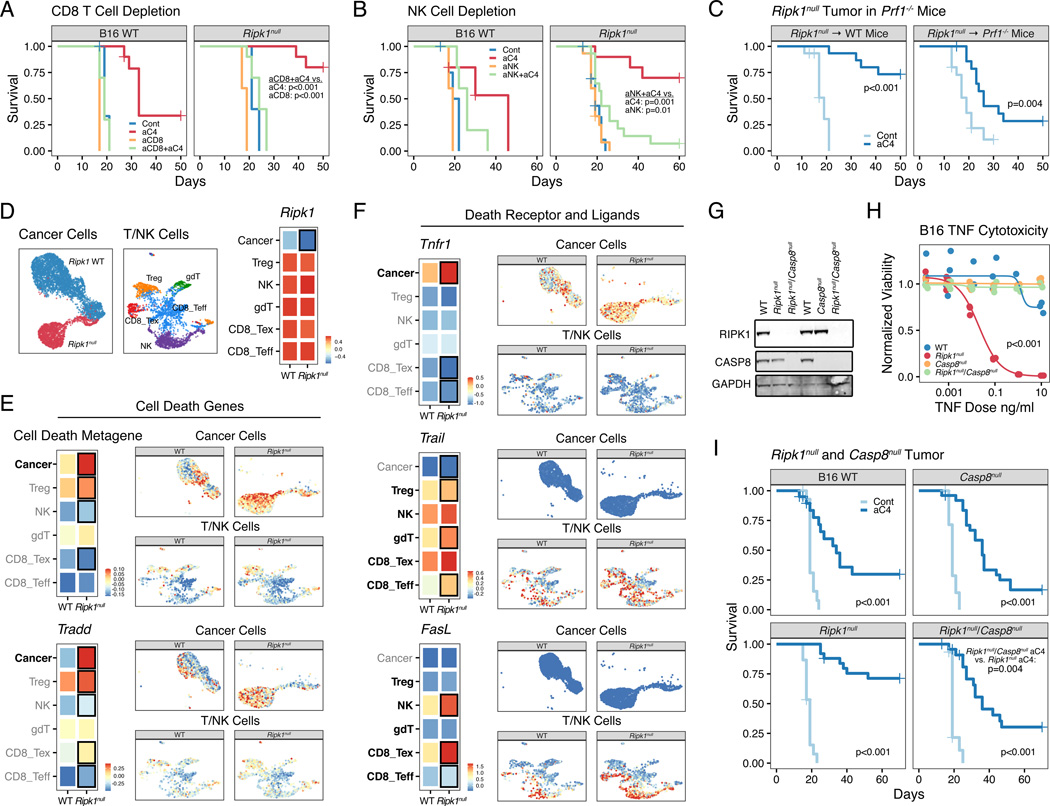
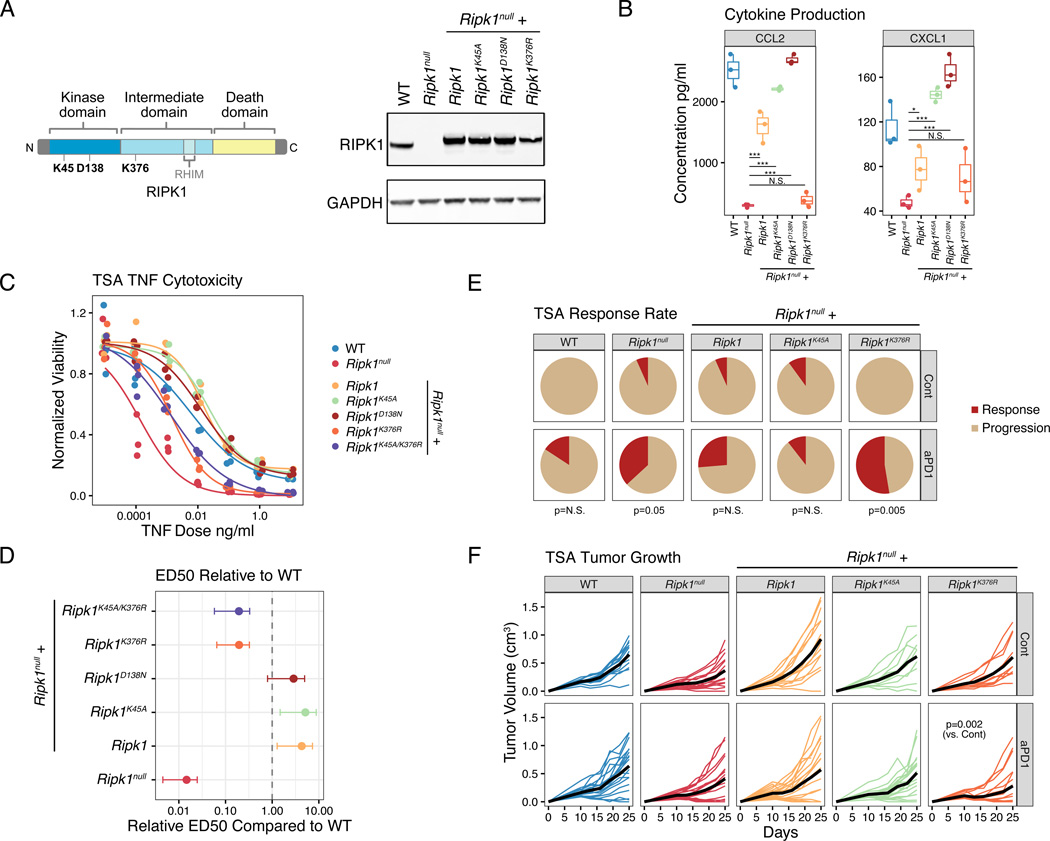
Interferon-gamma (IFN-γ) has pleiotropic effects on cancer immune checkpoint blockade (ICB), including roles in ICB resistance. We analyzed gene expression in ICB-sensitive versus ICB-resistant tumor cells and identified a strong association between interferon-mediated resistance and expression of Ripk1, a regulator of tumor necrosis factor (TNF) superfamily receptors. Genetic interaction screening revealed that in cancer cells, RIPK1 diverted TNF signaling through NF-κB and away from its role in cell death. This promoted an immunosuppressive chemokine program by cancer cells, enhanced cancer cell survival, and decreased infiltration of T and NK cells expressing TNF superfamily ligands. Deletion of RIPK1 in cancer cells compromised chemokine secretion, decreased ARG1+ suppressive myeloid cells linked to ICB failure in mice and humans, and improved ICB response driven by CASP8-killing and dependent on T and NK cells. RIPK1-mediated resistance required its ubiquitin scaffolding but not kinase function. Thus, cancer cells co-opt RIPK1 to promote cell-intrinsic and cell-extrinsic resistance to immunotherapy.
Copyright © 2022 Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests A.J.M. has received research funding from Merck. He is a scientific advisor for Takeda, H3Biomedicine, Xilio, and Related Sciences. A.J.M. is an inventor on patents related to the IFN pathway and an inventor on a filed patent related to modified CAR T cells. A.J.M. is a scientific founder for Dispatch Biotherapeutics.
Figures
Comment in
-
RIP(K)ing away immunotherapy resistance.Immunity. 2022 Apr 12;55(4):580-582. doi: 10.1016/j.immuni.2022.03.010. Immunity. 2022. PMID: 35417670
References
-
- Aaes TL, Kaczmarek A, Delvaeye T, De Craene B, De Koker S, Heyndrickx L, Delrue I, Taminau J, Wiernicki B, De Groote P, et al. (2016). Vaccination with Necroptotic Cancer Cells Induces Efficient Anti-tumor Immunity. Cell Rep. 15, 274–287. - PubMed
-
- Annibaldi A, and Meier P. (2018). Checkpoints in TNF-Induced Cell Death: Implications in Inflammation and Cancer. Trends Mol. Med. 24, 49–65. - PubMed
-
- Beck RJ, Slagter M, and Beltman JB (2019). Contact-Dependent Killing by Cytotoxic T Lymphocytes Is Insufficient for EL4 Tumor Regression In Vivo. Cancer Res. 79, 3406–3416. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
