Clinical and genetic features of a cohort of patients with MFN2-related neuropathy
- PMID: 35418194
- PMCID: PMC9008012
- DOI: 10.1038/s41598-022-10220-0
Clinical and genetic features of a cohort of patients with MFN2-related neuropathy
Abstract
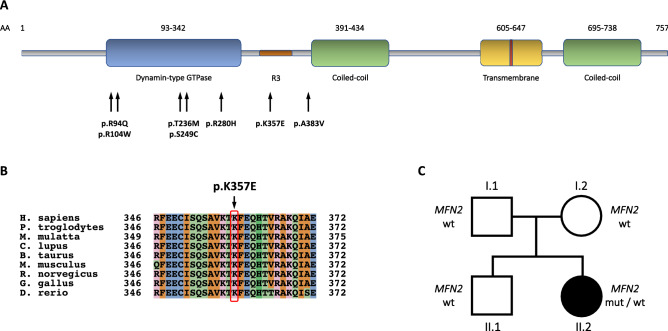
Charcot-Marie-Tooth disease type 2A (CMT2A) is a rare inherited axonal neuropathy caused by mutations in MFN2 gene, which encodes Mitofusin 2, a transmembrane protein of the outer mitochondrial membrane. We performed a cross-sectional analysis on thirteen patients carrying mutations in MFN2, from ten families, describing their clinical and genetic characteristics. Evaluated patients presented a variable age of onset and a wide phenotypic spectrum, with most patients presenting a severe phenotype. A novel heterozygous missense variant was detected, p.K357E. It is located at a highly conserved position and predicted as pathogenic by in silico tools. At a clinical level, the p.K357E carrier shows a severe sensorimotor axonal neuropathy. In conclusion, our work expands the genetic spectrum of CMT2A, disclosing a novel mutation and its related clinical effect, and provides a detailed description of the clinical features of a cohort of patients with MFN2 mutations. Obtaining a precise genetic diagnosis in affected families is crucial both for family planning and prenatal diagnosis, and in a therapeutic perspective, as we are entering the era of personalized therapy for genetic diseases.
© 2022. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures