

Somatic mutation rates scale with lifespan across mammals
- PMID: 35418684
- PMCID: PMC9021023
- DOI: 10.1038/s41586-022-04618-z
Somatic mutation rates scale with lifespan across mammals
Abstract
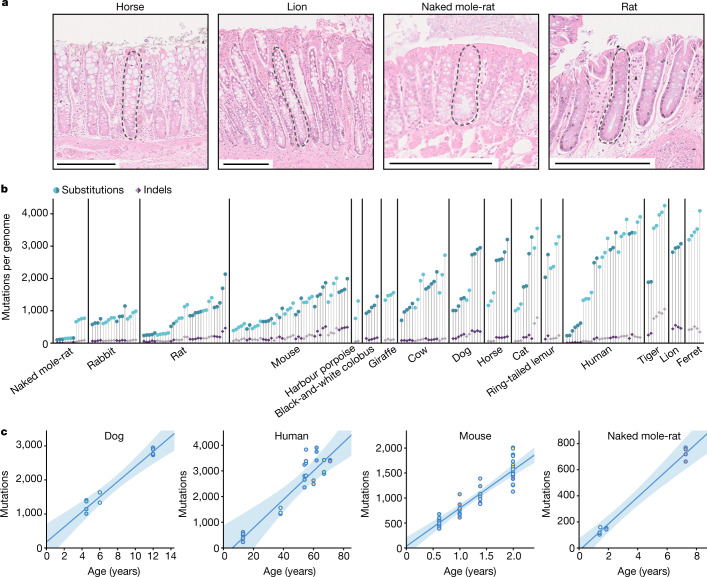
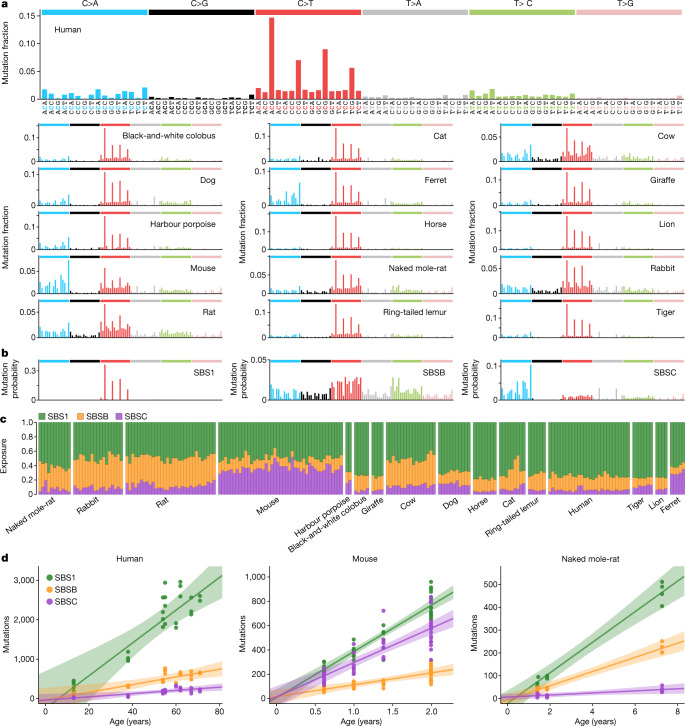
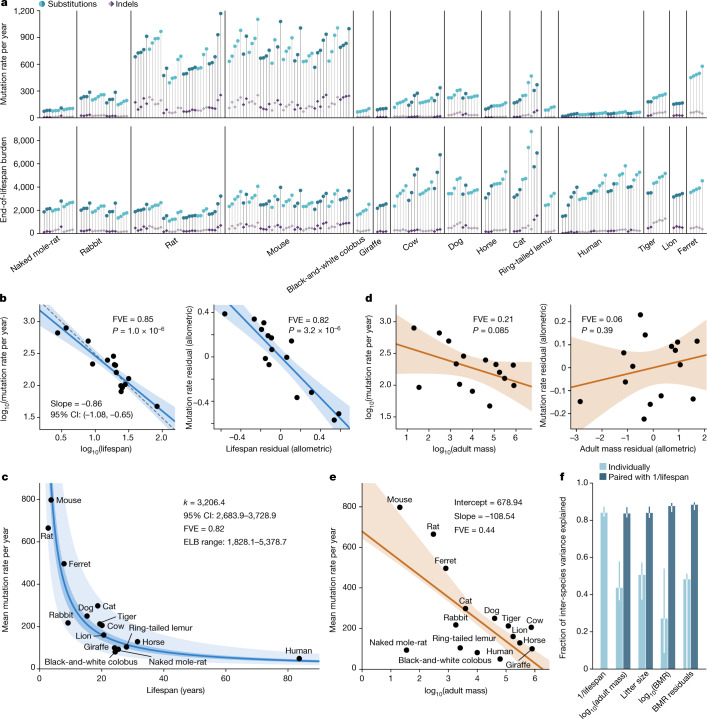
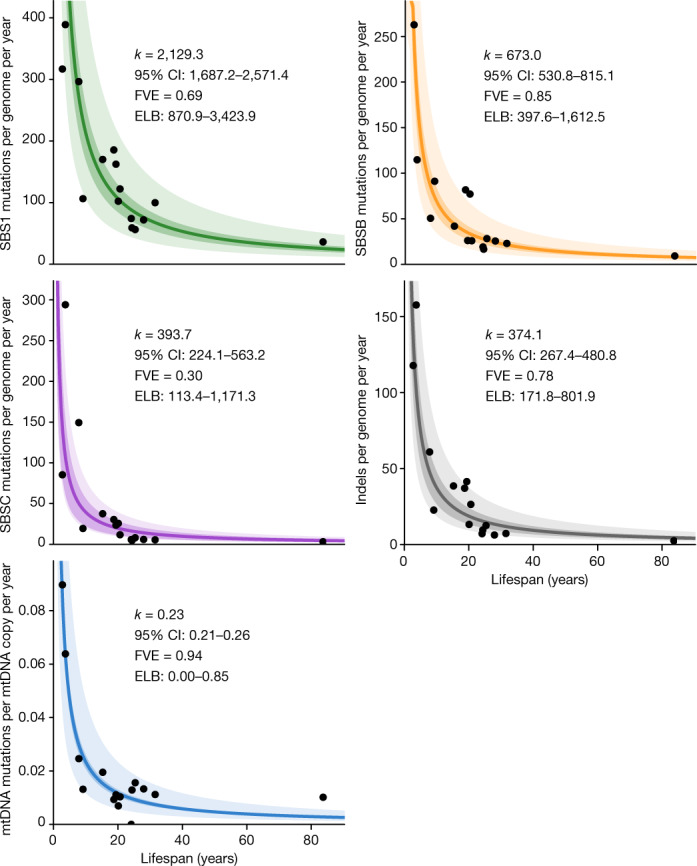
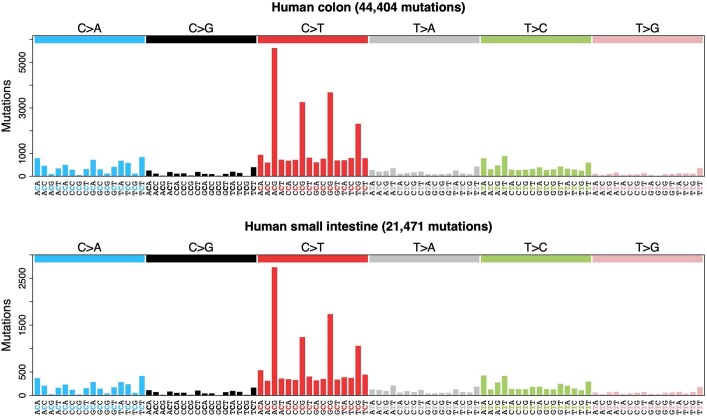
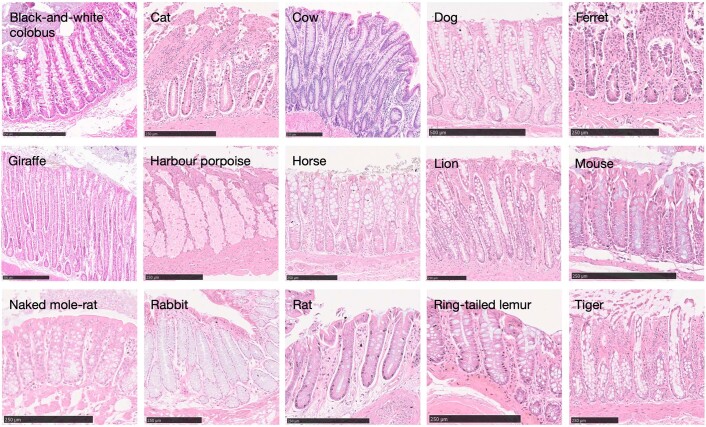
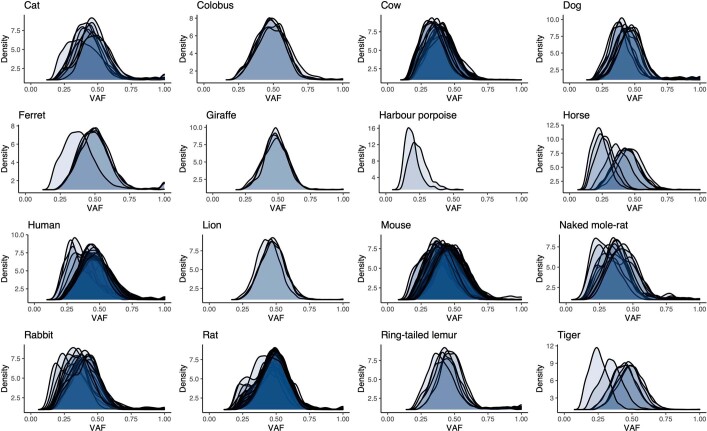
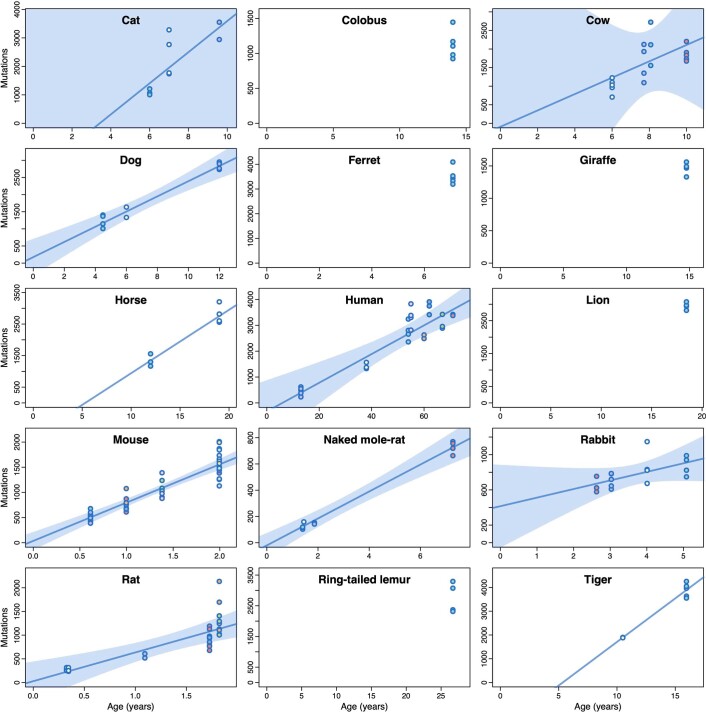
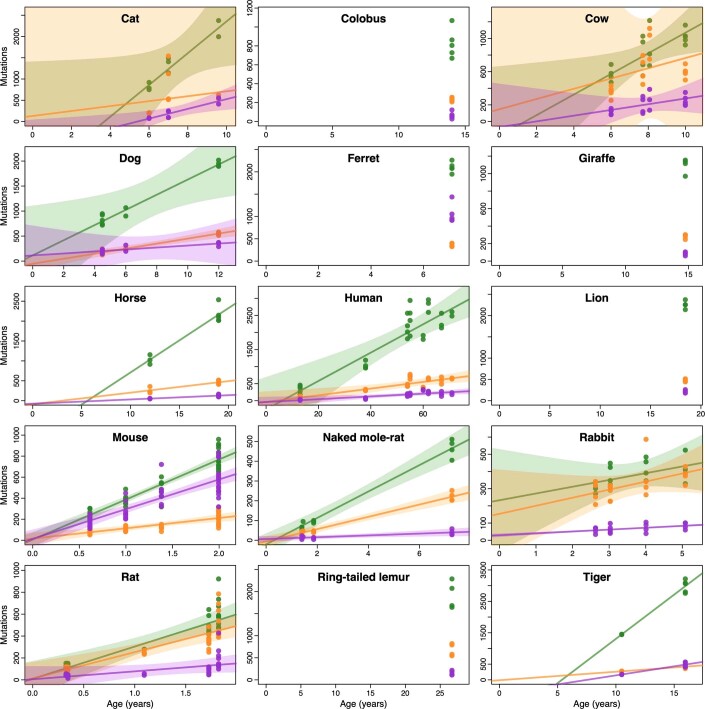
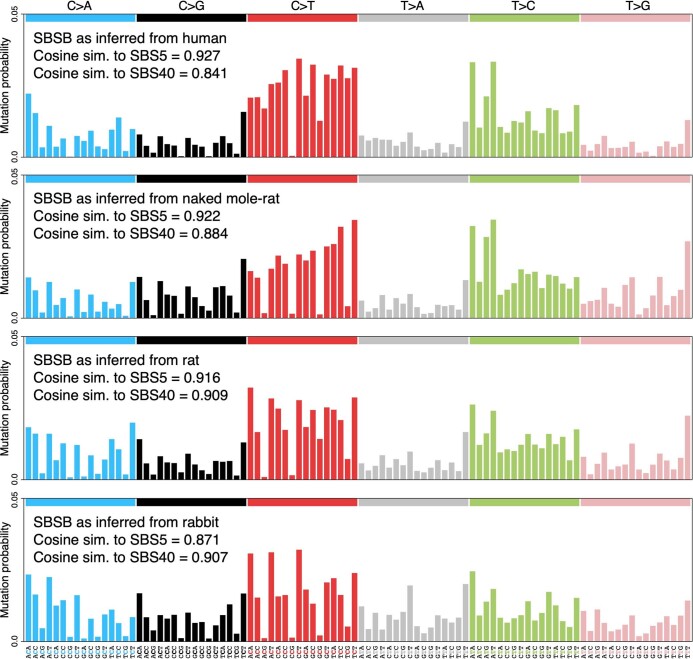
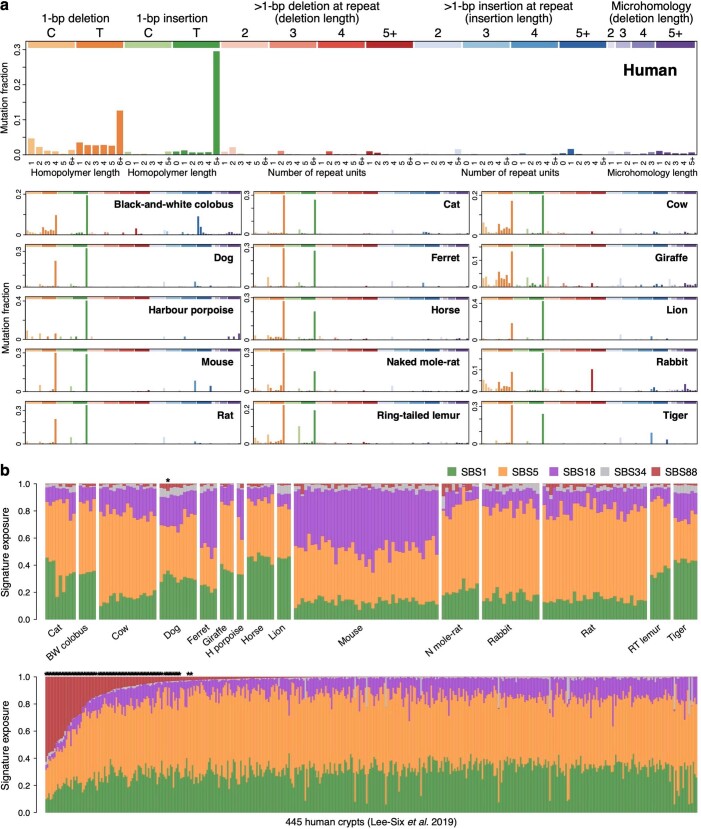
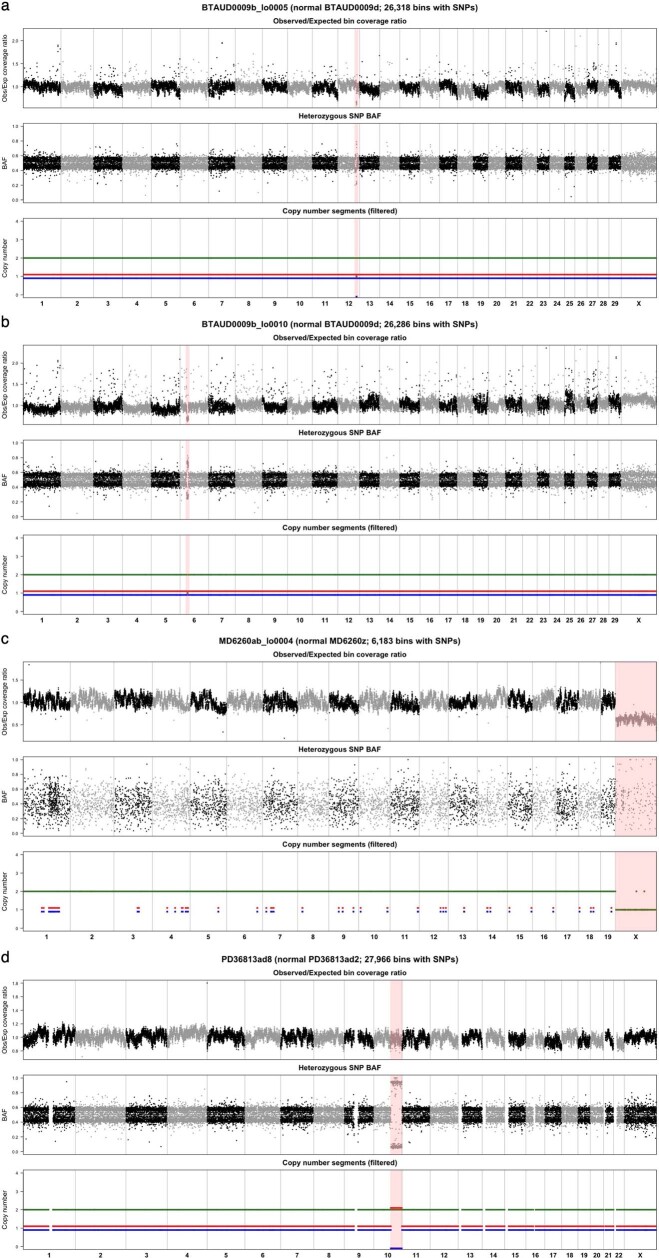
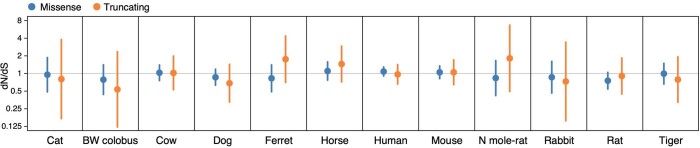
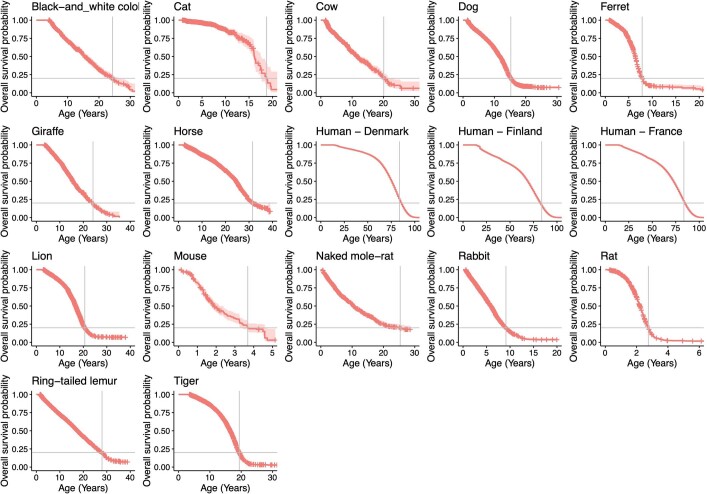
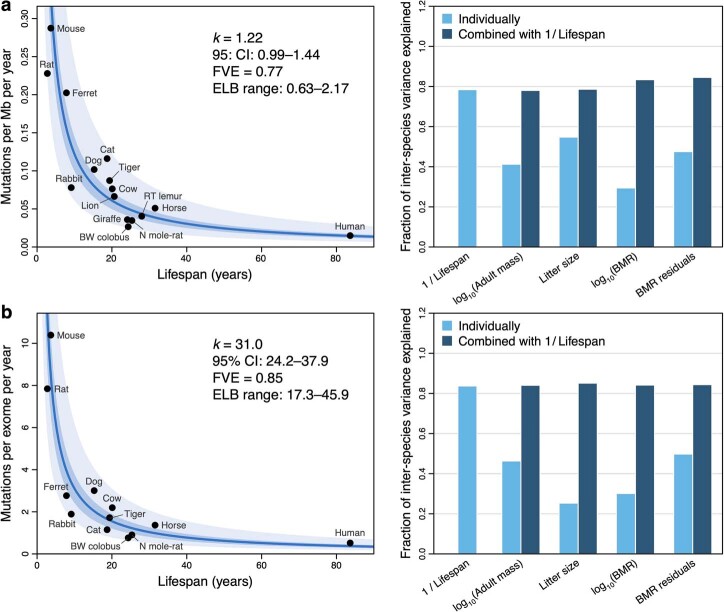
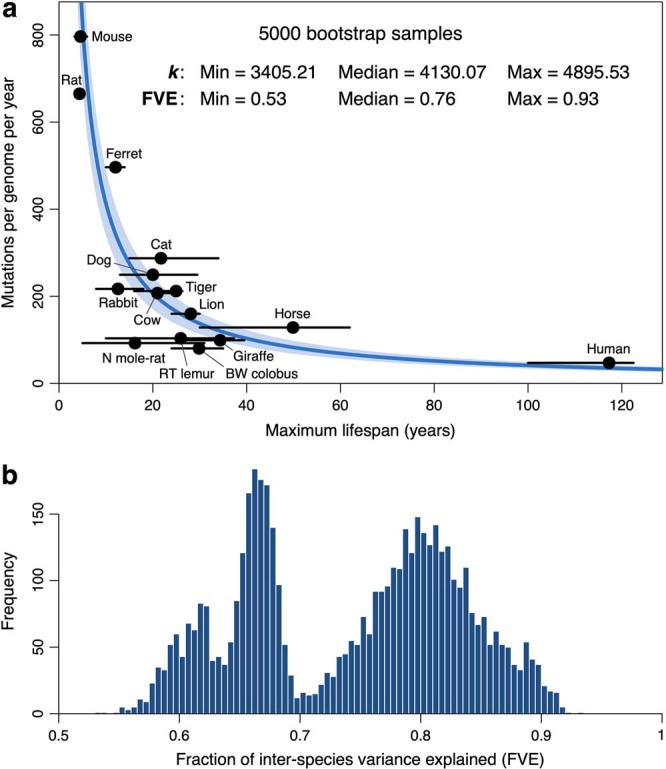
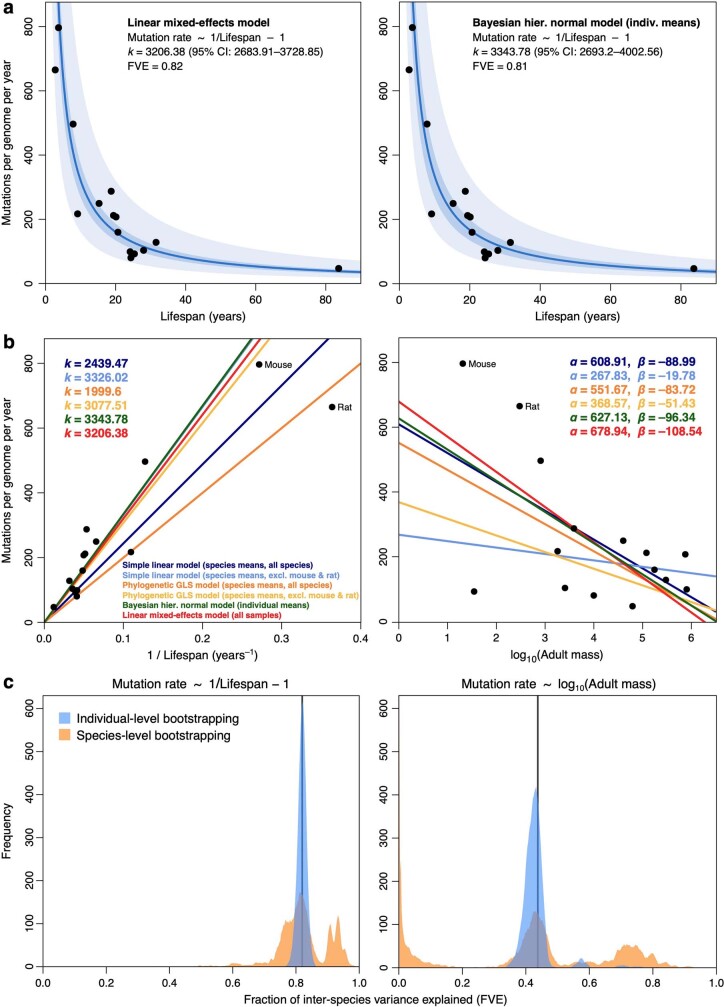
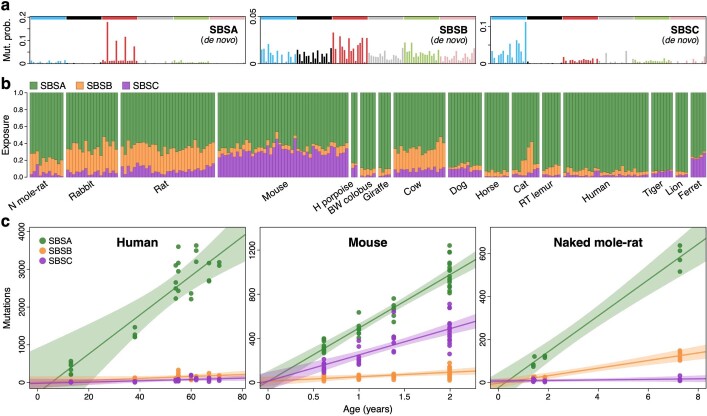
The rates and patterns of somatic mutation in normal tissues are largely unknown outside of humans1-7. Comparative analyses can shed light on the diversity of mutagenesis across species, and on long-standing hypotheses about the evolution of somatic mutation rates and their role in cancer and ageing. Here we performed whole-genome sequencing of 208 intestinal crypts from 56 individuals to study the landscape of somatic mutation across 16 mammalian species. We found that somatic mutagenesis was dominated by seemingly endogenous mutational processes in all species, including 5-methylcytosine deamination and oxidative damage. With some differences, mutational signatures in other species resembled those described in humans8, although the relative contribution of each signature varied across species. Notably, the somatic mutation rate per year varied greatly across species and exhibited a strong inverse relationship with species lifespan, with no other life-history trait studied showing a comparable association. Despite widely different life histories among the species we examined-including variation of around 30-fold in lifespan and around 40,000-fold in body mass-the somatic mutation burden at the end of lifespan varied only by a factor of around 3. These data unveil common mutational processes across mammals, and suggest that somatic mutation rates are evolutionarily constrained and may be a contributing factor in ageing.
© 2022. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures

Comment in
-
Mutational clocks tick differently across species.Nature. 2022 Apr;604(7906):435-436. doi: 10.1038/d41586-022-00976-w. Nature. 2022. PMID: 35418556 No abstract available.
-
Aging: Lifespan and the evolution of somatic mutation rates.Curr Biol. 2022 Jul 11;32(13):R753-R755. doi: 10.1016/j.cub.2022.06.002. Curr Biol. 2022. PMID: 35820389
-
Time's up: mutation rate and lifespan.Signal Transduct Target Ther. 2022 Aug 12;7(1):282. doi: 10.1038/s41392-022-01122-8. Signal Transduct Target Ther. 2022. PMID: 35961971 Free PMC article. No abstract available.
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
