

Inhibition of nonalcoholic fatty liver disease in mice by selective inhibition of mTORC1
- PMID: 35420934
- PMCID: PMC9811404
- DOI: 10.1126/science.abf8271
Inhibition of nonalcoholic fatty liver disease in mice by selective inhibition of mTORC1
Abstract
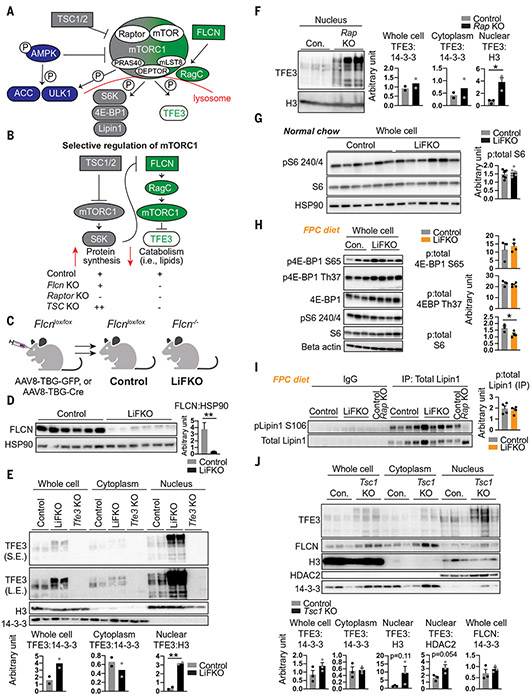
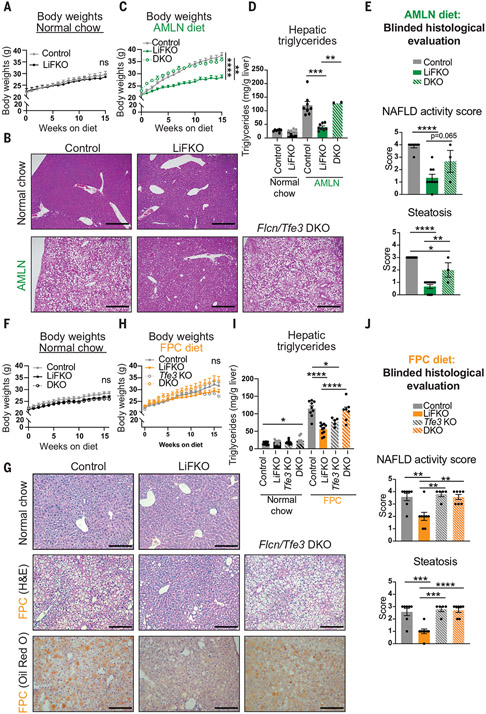
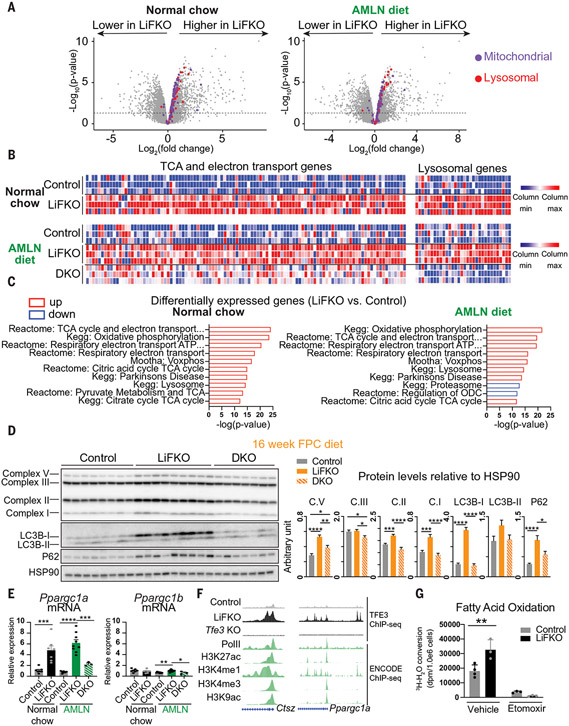
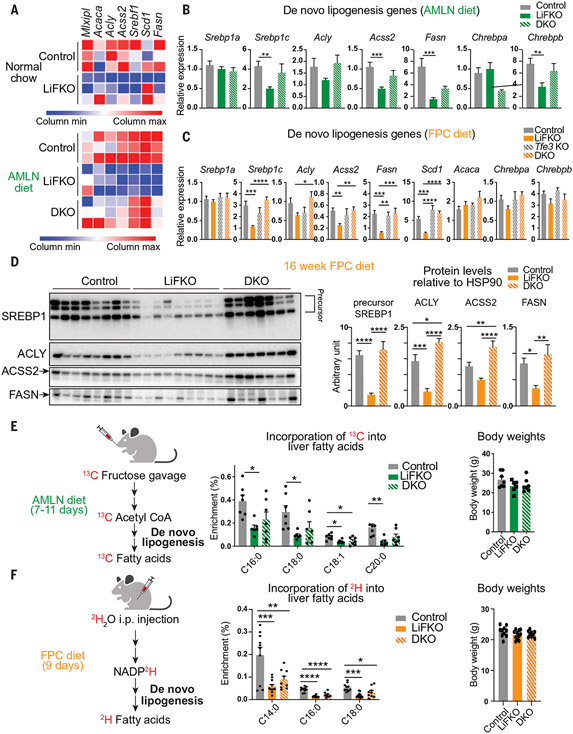
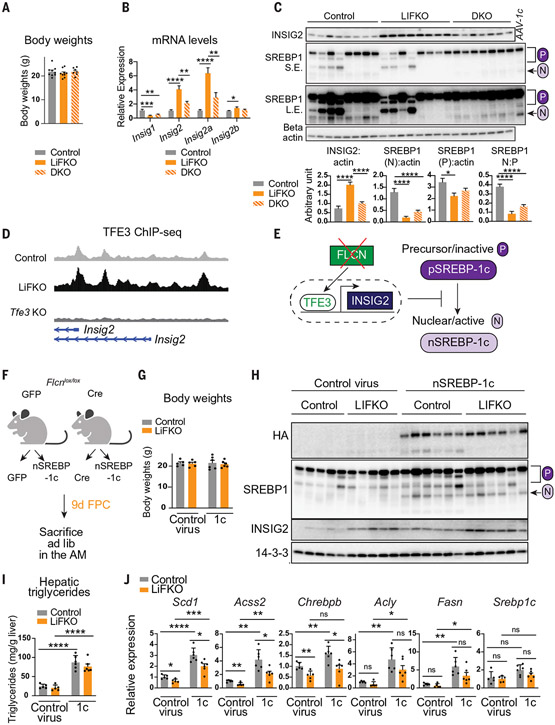
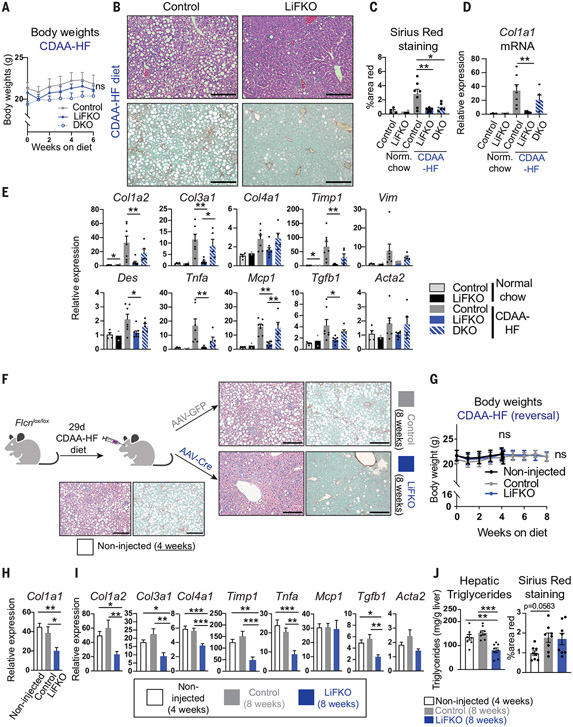
Nonalcoholic fatty liver disease (NAFLD) and nonalcoholic steatohepatitis (NASH) remain without effective therapies. The mechanistic target of rapamycin complex 1 (mTORC1) pathway is a potential therapeutic target, but conflicting interpretations have been proposed for how mTORC1 controls lipid homeostasis. We show that selective inhibition of mTORC1 signaling in mice, through deletion of the RagC/D guanosine triphosphatase-activating protein folliculin (FLCN), promotes activation of transcription factor E3 (TFE3) in the liver without affecting other mTORC1 targets and protects against NAFLD and NASH. Disease protection is mediated by TFE3, which both induces lipid consumption and suppresses anabolic lipogenesis. TFE3 inhibits lipogenesis by suppressing proteolytic processing and activation of sterol regulatory element-binding protein-1c (SREBP-1c) and by interacting with SREBP-1c on chromatin. Our data reconcile previously conflicting studies and identify selective inhibition of mTORC1 as a potential approach to treat NASH and NAFLD.
Figures
Comment in
-
Complex regulation of fatty liver disease.Science. 2022 Apr 15;376(6590):247-248. doi: 10.1126/science.abp8276. Epub 2022 Apr 14. Science. 2022. PMID: 35420931 Free PMC article.
-
Splitting Hairs: Folliculin Parts the Good From the Bad in mTORC1 Control Over Lipid Metabolism.Gastroenterology. 2022 Aug;163(2):529. doi: 10.1053/j.gastro.2022.06.008. Epub 2022 Jun 6. Gastroenterology. 2022. PMID: 35671806 No abstract available.
-
Targeting folliculin to selectively inhibit mTORC1: a promising strategy for treating nonalcoholic fatty liver disease.Signal Transduct Target Ther. 2022 Aug 9;7(1):277. doi: 10.1038/s41392-022-01111-x. Signal Transduct Target Ther. 2022. PMID: 35945199 Free PMC article. No abstract available.
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
