

Hmgcs2-mediated ketogenesis modulates high-fat diet-induced hepatosteatosis
- PMID: 35421611
- PMCID: PMC9039870
- DOI: 10.1016/j.molmet.2022.101494
Hmgcs2-mediated ketogenesis modulates high-fat diet-induced hepatosteatosis
Abstract
Objective: Aberrant ketogenesis is correlated with the degree of steatosis in non-alcoholic fatty liver disease (NAFLD) patients, and an inborn error of ketogenesis (mitochondrial HMG-CoA synthase deficiency) is commonly associated with the development of the fatty liver. Here we aimed to determine the impact of Hmgcs2-mediated ketogenesis and its modulations on the development and treatment of fatty liver disease.
Methods: Loss- and gain-of-ketogenic function models, achieved by Hmgcs2 knockout and overexpression, respectively, were utilized to investigate the role of ketogenesis in the hepatic lipid accumulation during postnatal development and in a high-fat diet-induced NAFLD mouse model.
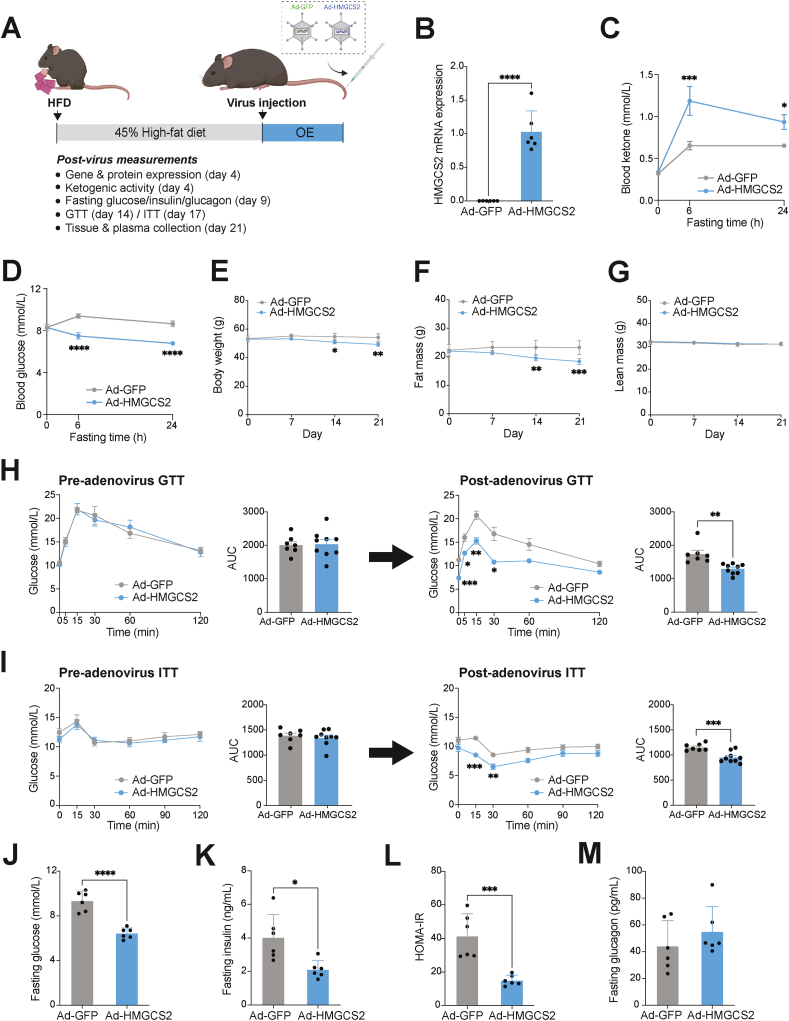
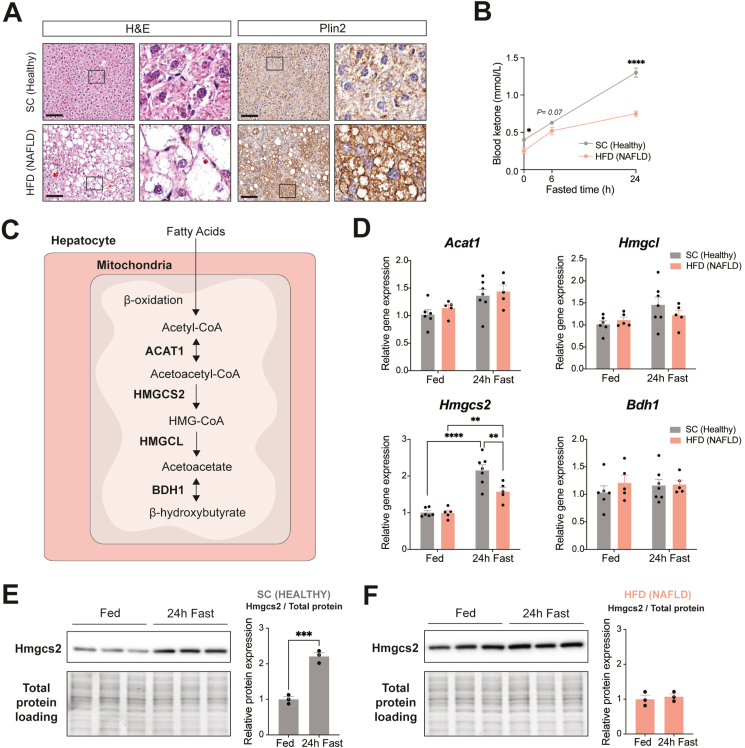
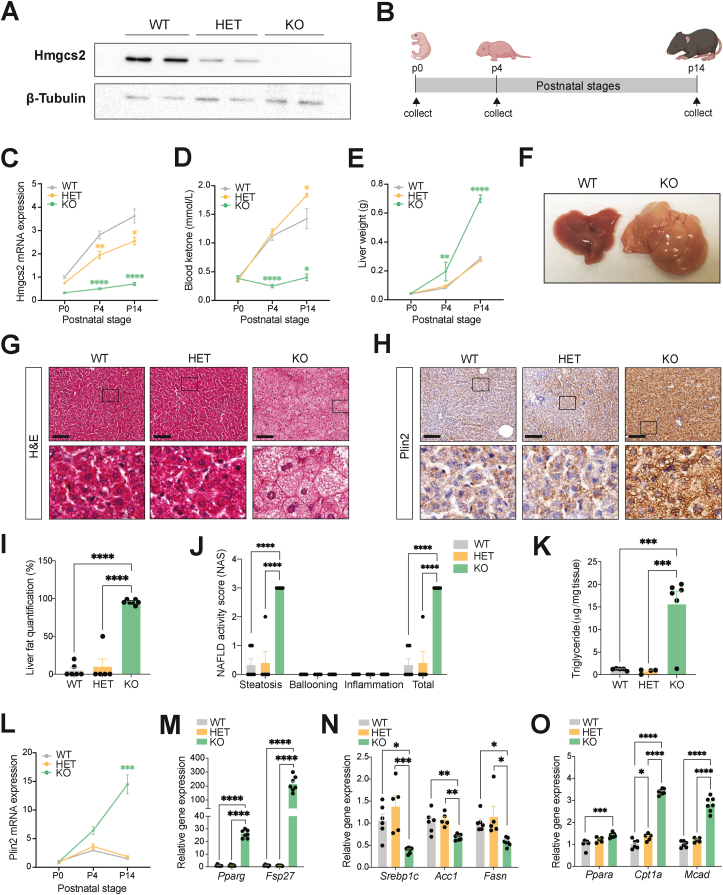
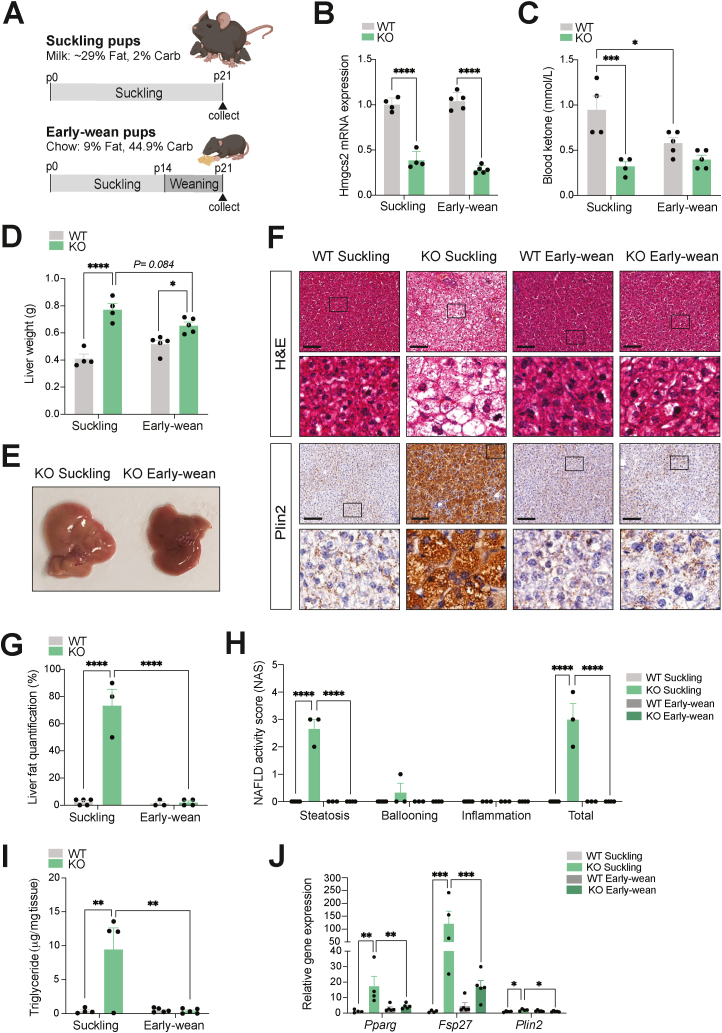
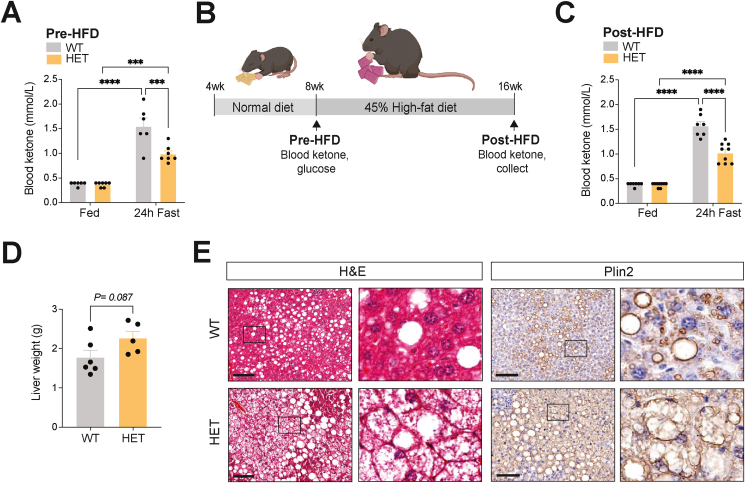
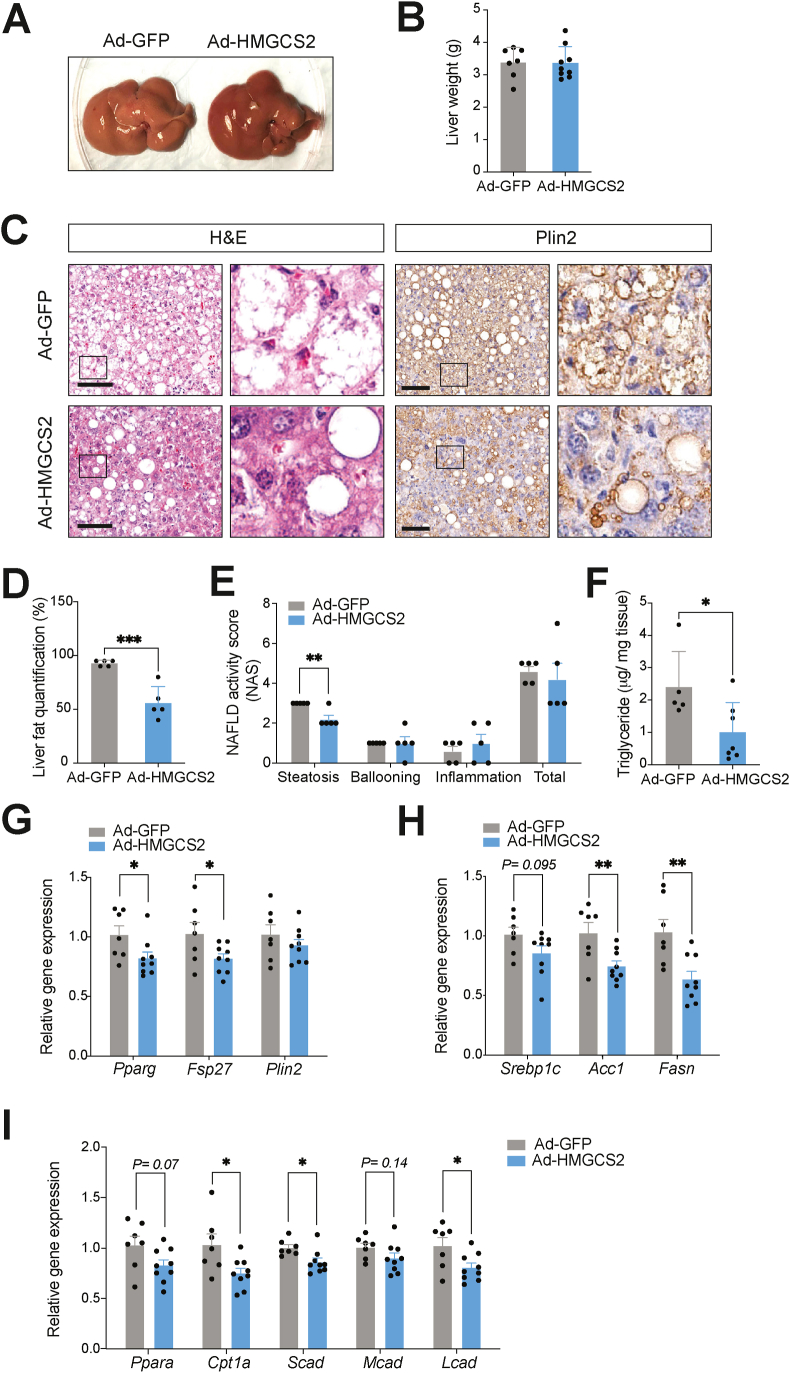
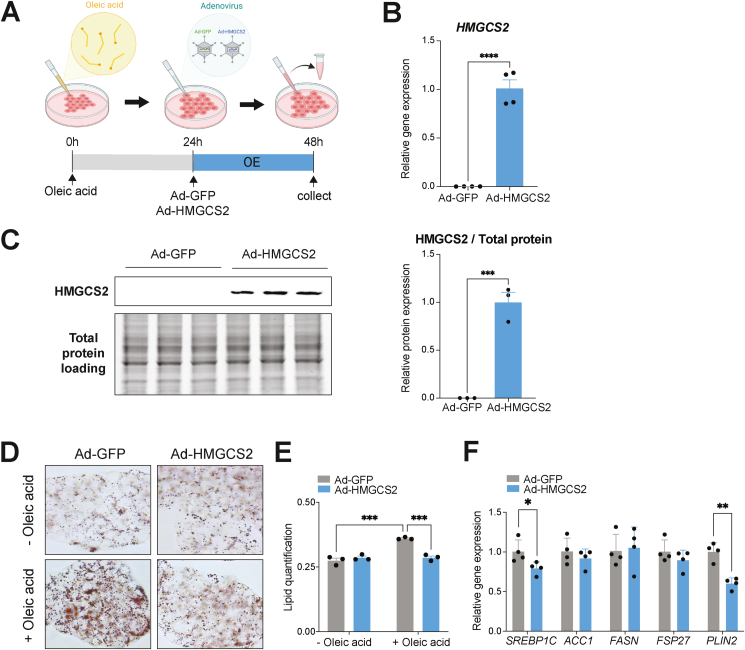
Results: Ketogenic function was decreased in NAFLD mice with a reduction in Hmgcs2 expression. Mice lacking Hmgcs2 developed spontaneous fatty liver phenotype during postnatal development, which was rescued by a shift to a low-fat dietary composition via early weaning. Hmgcs2 heterozygous adult mice, which exhibited lower ketogenic activity, were more susceptible to diet-induced NAFLD development, whereas HMGCS2 overexpression in NAFLD mice improved hepatosteatosis and glucose homeostasis.
Conclusions: Our study adds new knowledge to the field of ketone body metabolism and shows that Hmgcs2-mediated ketogenesis modulates hepatic lipid regulation under a fat-enriched nutritional environment. The regulation of hepatic ketogenesis may be a viable therapeutic strategy in the prevention and treatment of hepatosteatosis.
Keywords: Hmgcs2; Ketogenesis; Lipid accumulation; NAFLD.
Copyright © 2022 The Author(s). Published by Elsevier GmbH.. All rights reserved.
Figures
References
-
- Arshad T., Golabi P., Henry L., Younossi Z.M. Epidemiology of non-alcoholic fatty liver disease in north America. Current Pharmaceutical Design. 2020;26(10):993–997. - PubMed
-
- Makri E.S., Goulas A., Polyzos S.A. Sodium-glucose co-transporter 2 inhibitors in nonalcoholic fatty liver disease. European Journal of Pharmacology. 2021;907:174272. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
