A common framework of monocyte-derived macrophage activation
- PMID: 35427180
- PMCID: PMC12362712
- DOI: 10.1126/sciimmunol.abl7482
A common framework of monocyte-derived macrophage activation
Abstract
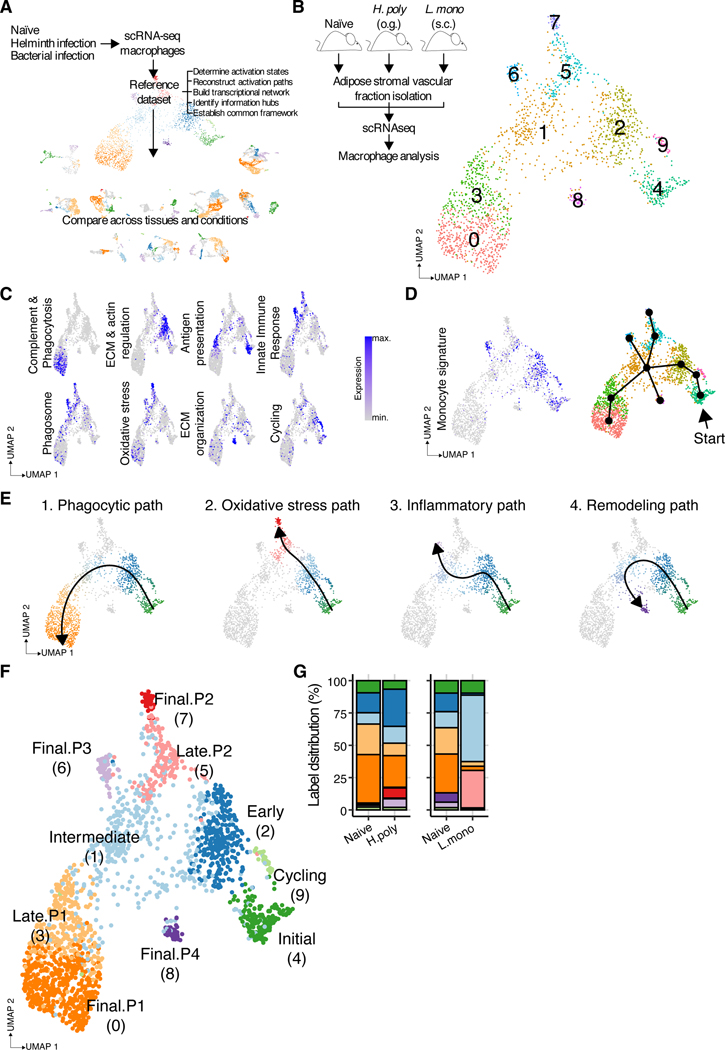
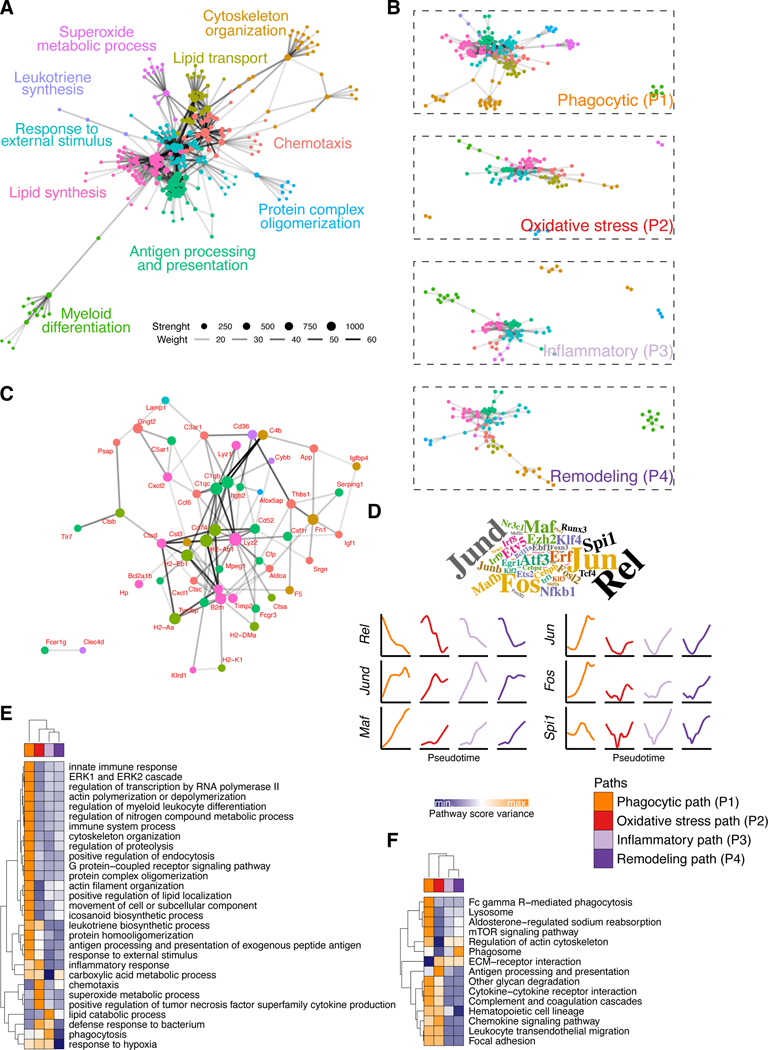
Macrophages populate every organ during homeostasis and disease, displaying features of tissue imprinting and heterogeneous activation. The disconnected picture of macrophage biology that has emerged from these observations is a barrier for integration across models or with in vitro macrophage activation paradigms. We set out to contextualize macrophage heterogeneity across mouse tissues and inflammatory conditions, specifically aiming to define a common framework of macrophage activation. We built a predictive model with which we mapped the activation of macrophages across 12 tissues and 25 biological conditions, finding a notable commonality and finite number of transcriptional profiles, in particular among infiltrating macrophages, which we modeled as defined stages along four conserved activation paths. These activation paths include a "phagocytic" regulatory path, an "inflammatory" cytokine-producing path, an "oxidative stress" antimicrobial path, or a "remodeling" extracellular matrix deposition path. We verified this model with adoptive cell transfer experiments and identified transient RELMɑ expression as a feature of monocyte-derived macrophage tissue engraftment. We propose that this integrative approach of macrophage classification allows the establishment of a common predictive framework of monocyte-derived macrophage activation in inflammation and homeostasis.
Conflict of interest statement
Figures





Comment in
-
Redefining inflammatory macrophage phenotypes across stages and tissues by single-cell transcriptomics.Sci Immunol. 2022 Apr 15;7(70):eabo4652. doi: 10.1126/sciimmunol.abo4652. Epub 2022 Apr 15. Sci Immunol. 2022. PMID: 35427177
References
-
- Gautier EL, Shay T, Miller J, Greter M, Jakubzick C, Ivanov S, Helft J, Chow A, Elpek KG, Gordonov S, Mazloom AR, Ma’ayan A, Chua W-J, Hansen TH, Turley SJ, Merad M, Randolph GJ, Immunological Genome Consortium, Gene-expression profiles and transcriptional regulatory pathways that underlie the identity and diversity of mouse tissue macrophages. Nat. Immunol 13, 1118–1128 (2012). - PMC - PubMed
-
- Hoeffel G, Ginhoux F, Fetal monocytes and the origins of tissue-resident macrophages. Cell. Immunol 330, 5–15 (2018). - PubMed
-
- Mass E, Ballesteros I, Farlik M, Halbritter F, Günther P, Crozet L, Jacome-Galarza CE, Händler K, Klughammer J, Kobayashi Y, Gomez-Perdiguero E, Schultze JL, Beyer M, Bock C, Geissmann F, Specification of tissue-resident macrophages during organogenesis. Science. 353 (2016), doi: 10.1126/science.aaf4238. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases