

Expanding the clinical-pathological and genetic spectrum of RYR1-related congenital myopathies with cores and minicores: an Italian population study
- PMID: 35428369
- PMCID: PMC9013059
- DOI: 10.1186/s40478-022-01357-0
Expanding the clinical-pathological and genetic spectrum of RYR1-related congenital myopathies with cores and minicores: an Italian population study
Abstract
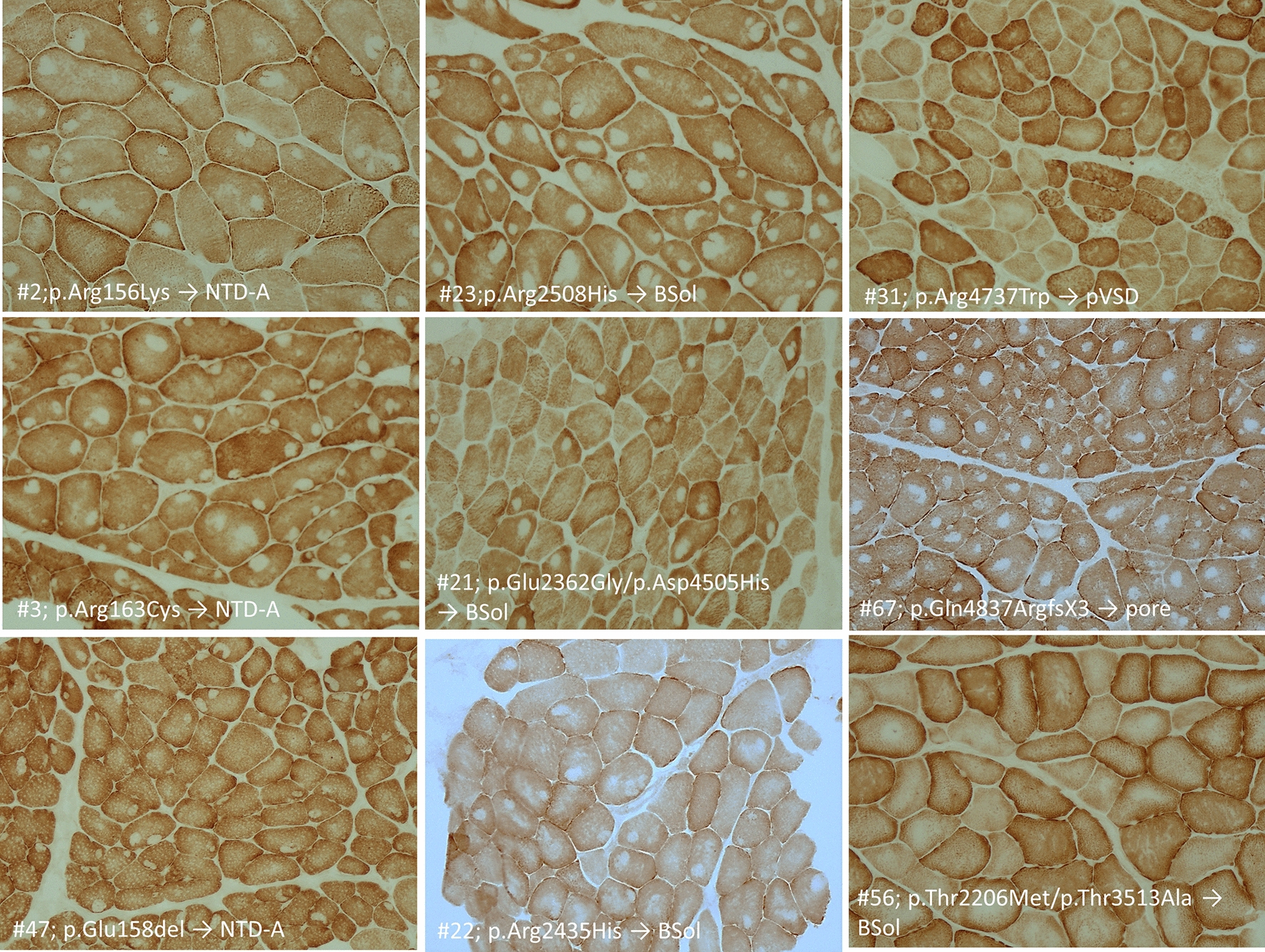
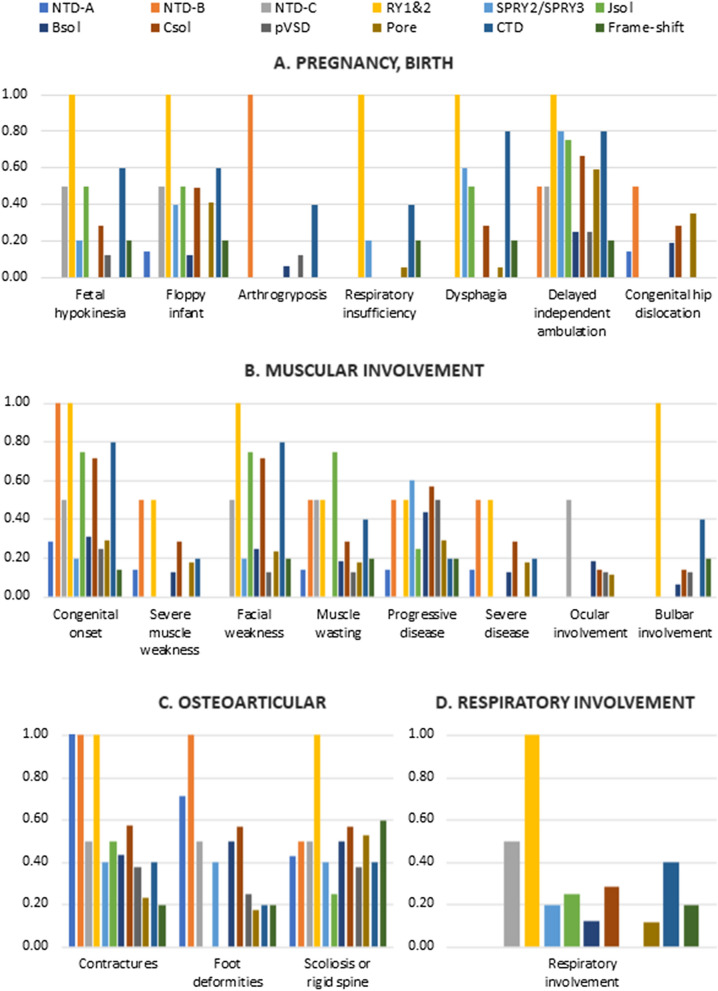
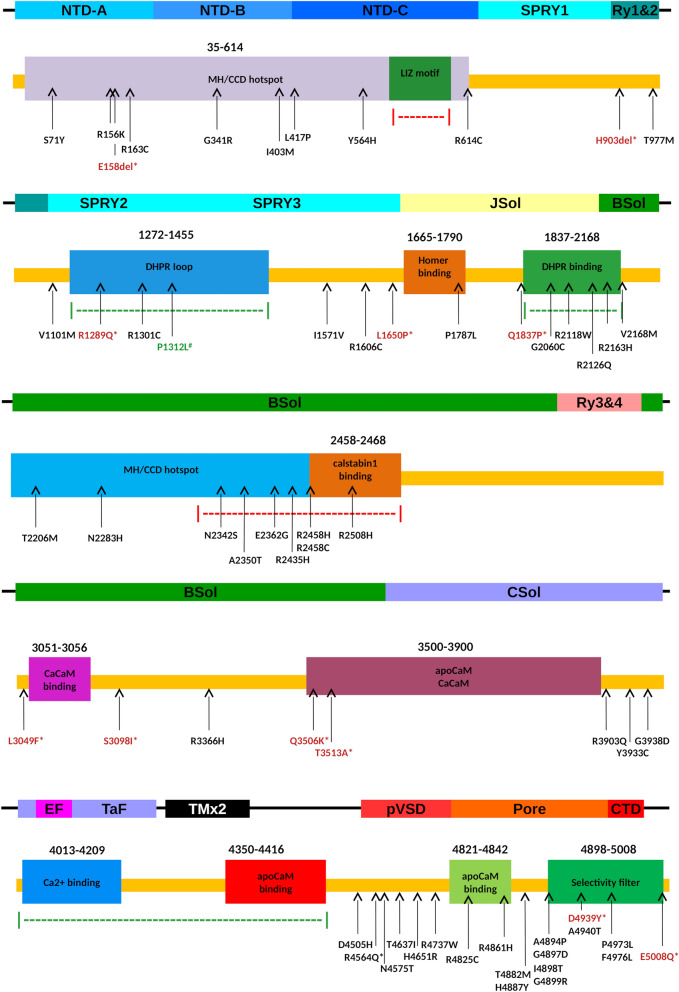
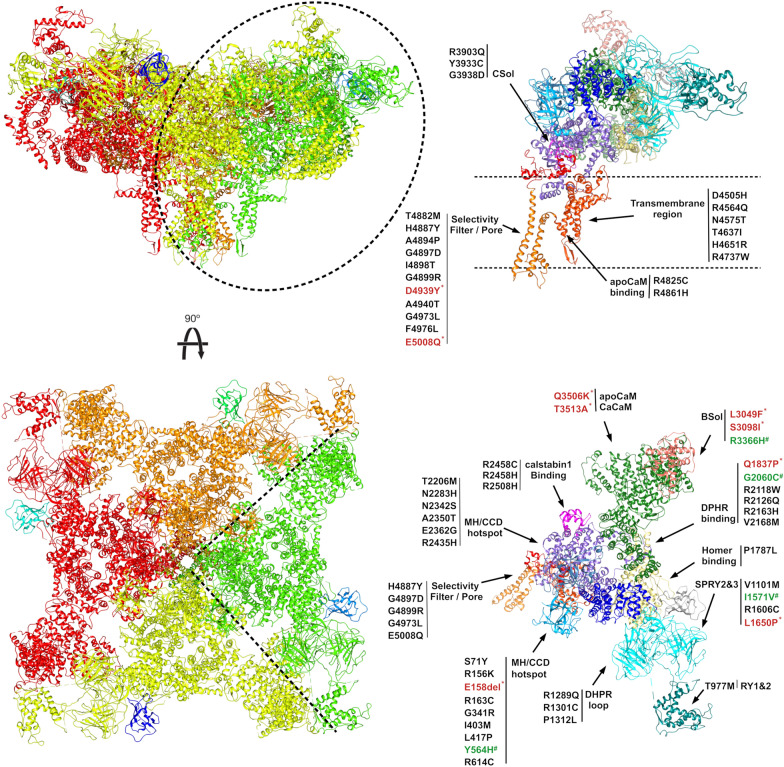
Mutations in the RYR1 gene, encoding ryanodine receptor 1 (RyR1), are a well-known cause of Central Core Disease (CCD) and Multi-minicore Disease (MmD). We screened a cohort of 153 patients carrying an histopathological diagnosis of core myopathy (cores and minicores) for RYR1 mutation. At least one RYR1 mutation was identified in 69 of them and these patients were further studied. Clinical and histopathological features were collected. Clinical phenotype was highly heterogeneous ranging from asymptomatic or paucisymptomatic hyperCKemia to severe muscle weakness and skeletal deformity with loss of ambulation. Sixty-eight RYR1 mutations, generally missense, were identified, of which 16 were novel. The combined analysis of the clinical presentation, disease progression and the structural bioinformatic analyses of RYR1 allowed to associate some phenotypes to mutations in specific domains. In addition, this study highlighted the structural bioinformatics potential in the prediction of the pathogenicity of RYR1 mutations. Further improvement in the comprehension of genotype-phenotype relationship of core myopathies can be expected in the next future: the actual lack of the human RyR1 crystal structure paired with the presence of large intrinsically disordered regions in RyR1, and the frequent presence of more than one RYR1 mutation in core myopathy patients, require designing novel investigation strategies to completely address RyR1 mutation effect.
Keywords: Central core disease; Genotype–phenotype correlations; Multi-minicore disease; Neuromuscular disorder; Protein modelling; RYR1-related myopathies.
© 2022. The Author(s).
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
References
-
- De Cauwer H, Heytens L, Martin J-J (2002) Workshop report of the 89th ENMC International Workshop: Central Core Disease, 19th-20th January 2001, Hilversum, The Netherlands. Neuromuscul Disord [Internet] 12:588–95. 10.1016/S0960-8966(02)00002-0 - PubMed
