

Recent Advances in the Modeling of Alzheimer's Disease
- PMID: 35431779
- PMCID: PMC9009508
- DOI: 10.3389/fnins.2022.807473
Recent Advances in the Modeling of Alzheimer's Disease
Abstract
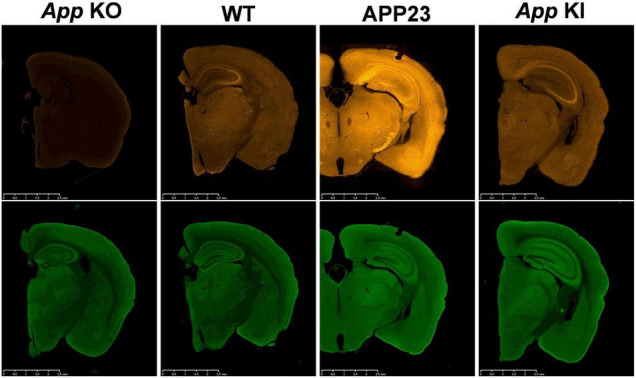
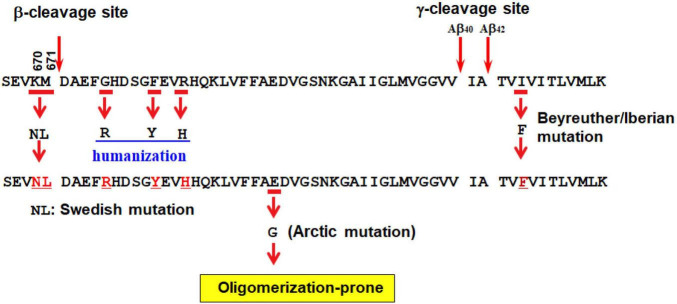
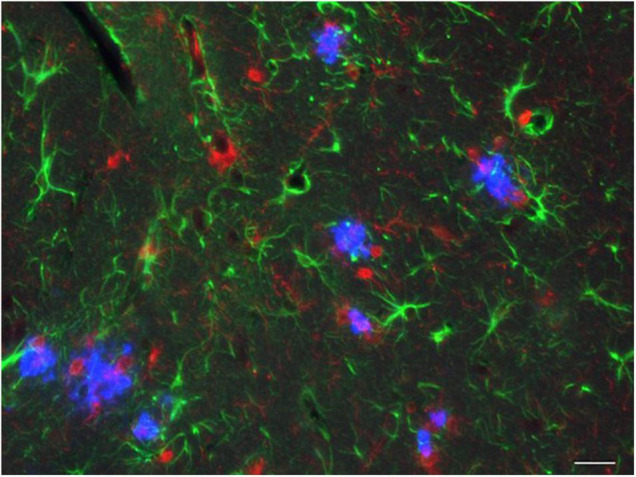
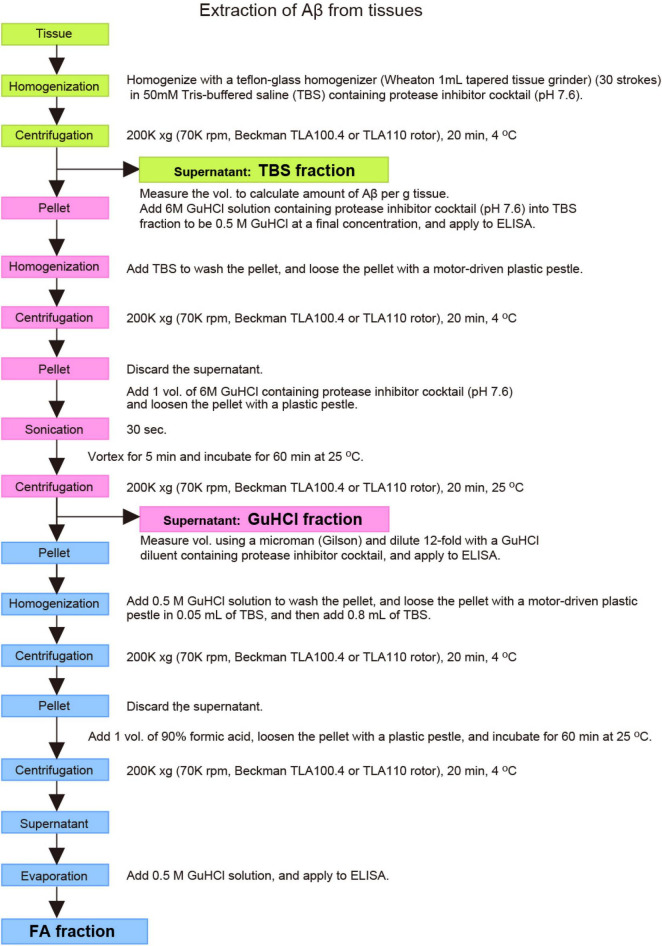
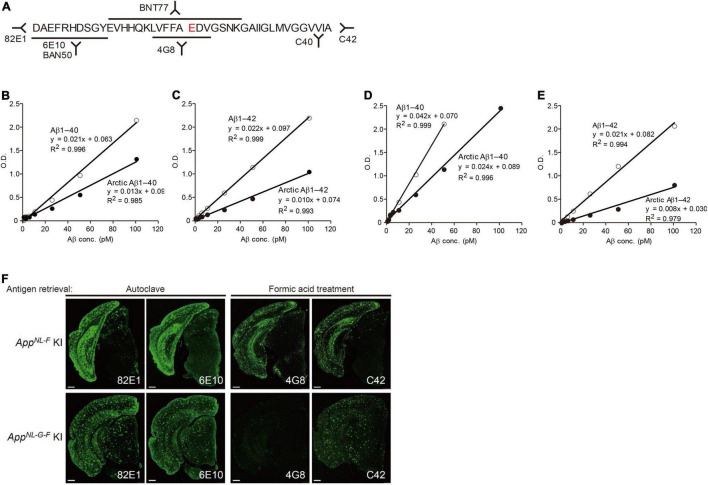
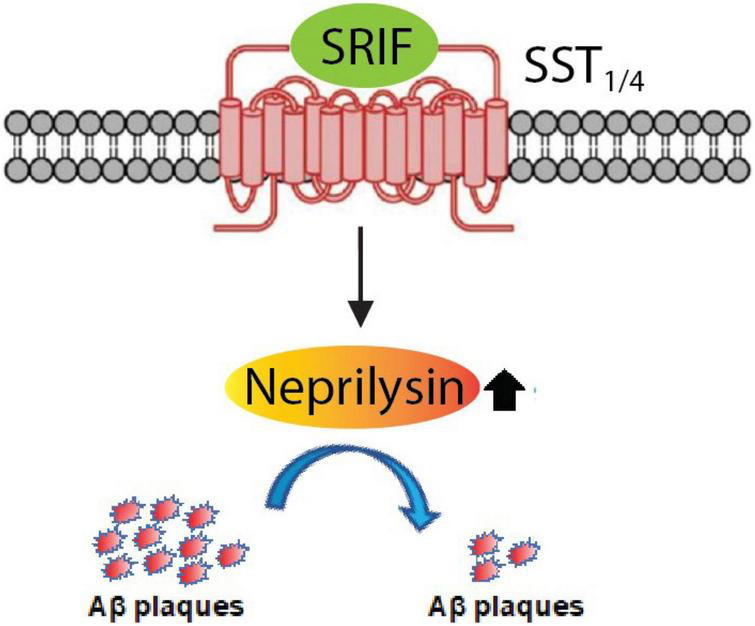
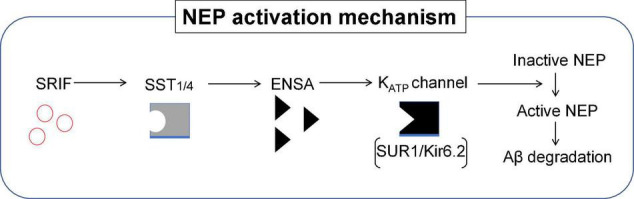
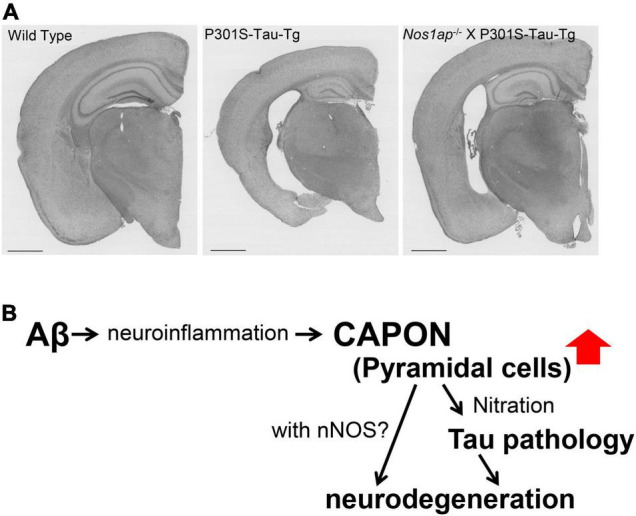
Since 1995, more than 100 transgenic (Tg) mouse models of Alzheimer's disease (AD) have been generated in which mutant amyloid precursor protein (APP) or APP/presenilin 1 (PS1) cDNA is overexpressed ( 1st generation models ). Although many of these models successfully recapitulate major pathological hallmarks of the disease such as amyloid β peptide (Aβ) deposition and neuroinflammation, they have suffered from artificial phenotypes in the form of overproduced or mislocalized APP/PS1 and their functional fragments, as well as calpastatin deficiency-induced early lethality, calpain activation, neuronal cell death without tau pathology, endoplasmic reticulum stresses, and inflammasome involvement. Such artifacts bring two important uncertainties into play, these being (1) why the artifacts arise, and (2) how they affect the interpretation of experimental results. In addition, destruction of endogenous gene loci in some Tg lines by transgenes has been reported. To overcome these concerns, single App knock-in mouse models harboring the Swedish and Beyreuther/Iberian mutations with or without the Arctic mutation (AppNL-G-F and AppNL-F mice) were developed ( 2nd generation models ). While these models are interesting given that they exhibit Aβ pathology, neuroinflammation, and cognitive impairment in an age-dependent manner, the model with the Artic mutation, which exhibits an extensive pathology as early as 6 months of age, is not suitable for investigating Aβ metabolism and clearance because the Aβ in this model is resistant to proteolytic degradation and is therefore prone to aggregation. Moreover, it cannot be used for preclinical immunotherapy studies owing to the discrete affinity it shows for anti-Aβ antibodies. The weakness of the latter model (without the Arctic mutation) is that the pathology may require up to 18 months before it becomes sufficiently apparent for experimental investigation. Nevertheless, this model was successfully applied to modulating Aβ pathology by genome editing, to revealing the differential roles of neprilysin and insulin-degrading enzyme in Aβ metabolism, and to identifying somatostatin receptor subtypes involved in Aβ degradation by neprilysin. In addition to discussing these issues, we also provide here a technical guide for the application of App knock-in mice to AD research. Subsequently, a new double knock-in line carrying the AppNL-F and Psen1 P117L/WT mutations was generated, the pathogenic effect of which was found to be synergistic. A characteristic of this 3rd generation model is that it exhibits more cored plaque pathology and neuroinflammation than the AppNL-G-F line, and thus is more suitable for preclinical studies of disease-modifying medications targeting Aβ. Furthermore, a derivative AppG-F line devoid of Swedish mutations which can be utilized for preclinical studies of β-secretase modifier(s) was recently created. In addition, we introduce a new model of cerebral amyloid angiopathy that may be useful for analyzing amyloid-related imaging abnormalities that can be caused by anti-Aβ immunotherapy. Use of the App knock-in mice also led to identification of the α-endosulfine-K ATP channel pathway as components of the somatostatin-evoked physiological mechanisms that reduce Aβ deposition via the activation of neprilysin. Such advances have provided new insights for the prevention and treatment of preclinical AD. Because tau pathology plays an essential role in AD pathogenesis, knock-in mice with human tau wherein the entire murine Mapt gene has been humanized were generated. Using these mice, the carboxy-terminal PDZ ligand of neuronal nitric oxide synthase (CAPON) was discovered as a mediator linking tau pathology to neurodegeneration and showed that tau humanization promoted pathological tau propagation. Finally, we describe and discuss the current status of mutant human tau knock-in mice and a non-human primate model of AD that we have successfully created.
Keywords: Alzheimer’s disease; amyloid – beta; amyloidosis; mouse model; non-human primate (NHP); somatostatin; tau propagation.
Copyright © 2022 Sasaguri, Hashimoto, Watamura, Sato, Takamura, Nagata, Tsubuki, Ohshima, Yoshiki, Sato, Kumita, Sasaki, Kitazume, Nilsson, Winblad, Saito, Iwata and Saido.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures




References
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous