AUTS2 Syndrome: Molecular Mechanisms and Model Systems
- PMID: 35431798
- PMCID: PMC9008325
- DOI: 10.3389/fnmol.2022.858582
AUTS2 Syndrome: Molecular Mechanisms and Model Systems
Abstract
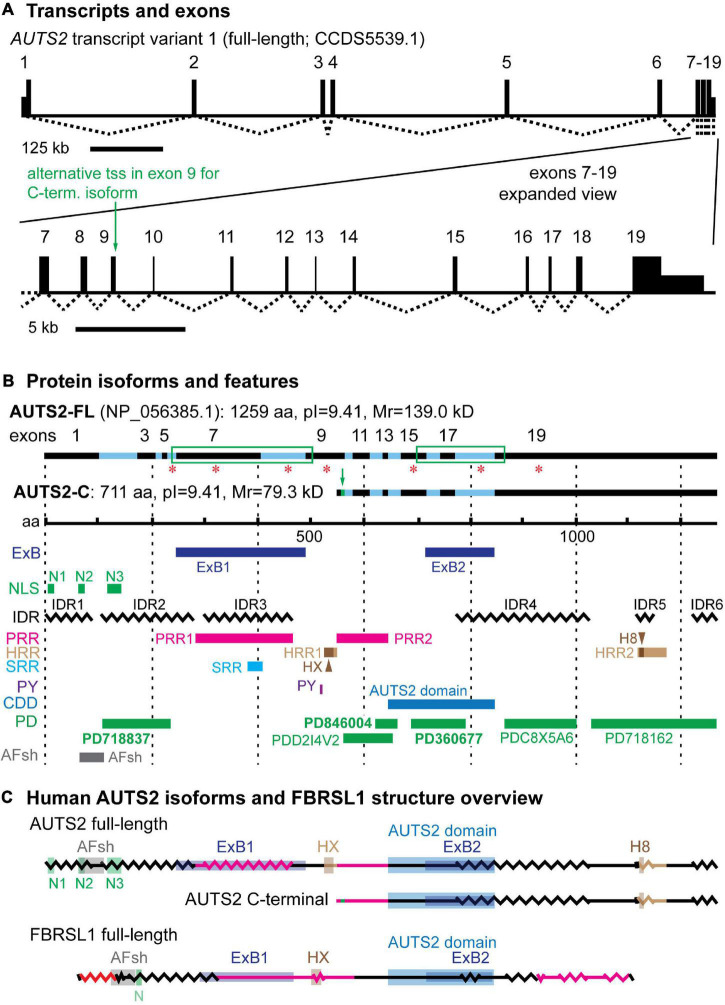
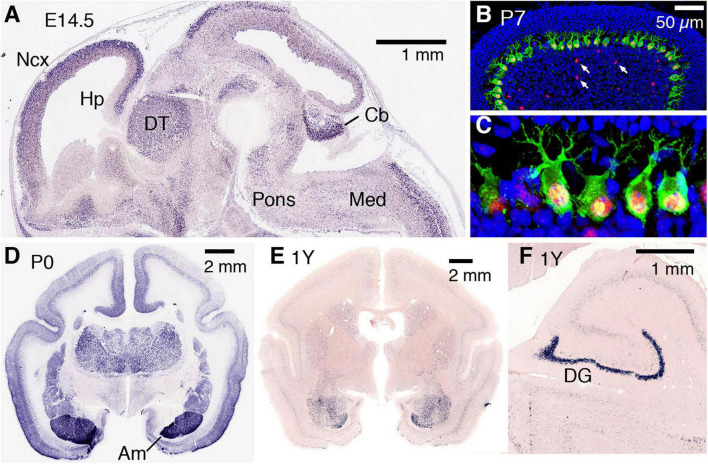
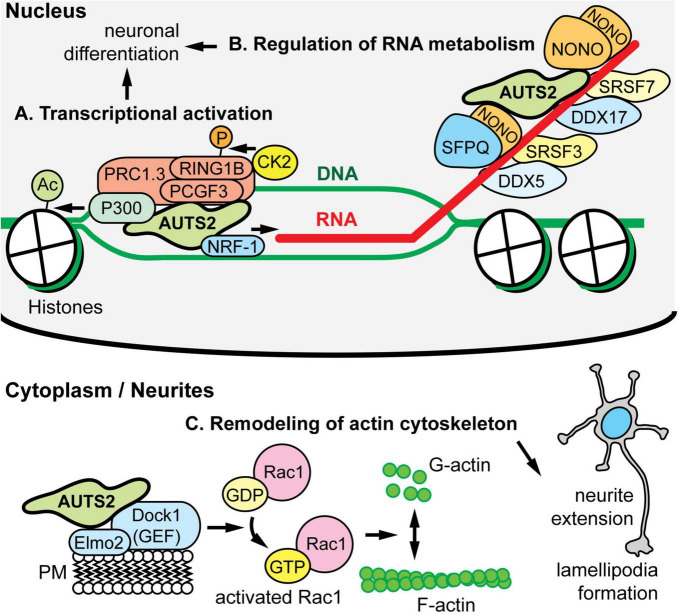
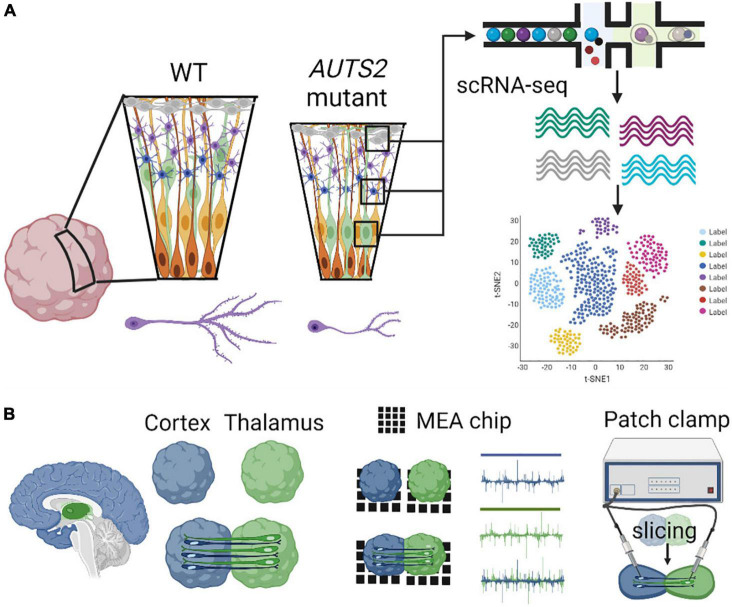
AUTS2 syndrome is a genetic disorder that causes intellectual disability, microcephaly, and other phenotypes. Syndrome severity is worse when mutations involve 3' regions (exons 9-19) of the AUTS2 gene. Human AUTS2 protein has two major isoforms, full-length (1259 aa) and C-terminal (711 aa), the latter produced from an alternative transcription start site in exon 9. Structurally, AUTS2 contains the putative "AUTS2 domain" (∼200 aa) conserved among AUTS2 and its ohnologs, fibrosin, and fibrosin-like-1. Also, AUTS2 contains extensive low-complexity sequences and intrinsically disordered regions, features typical of RNA-binding proteins. During development, AUTS2 is expressed by specific progenitor cell and neuron types, including pyramidal neurons and Purkinje cells. AUTS2 localizes mainly in cell nuclei, where it regulates transcription and RNA metabolism. Some studies have detected AUTS2 in neurites, where it may regulate cytoskeletal dynamics. Neurodevelopmental functions of AUTS2 have been studied in diverse model systems. In zebrafish, auts2a morphants displayed microcephaly. In mice, excision of different Auts2 exons (7, 8, or 15) caused distinct phenotypes, variously including neonatal breathing abnormalities, cerebellar hypoplasia, dentate gyrus hypoplasia, EEG abnormalities, and behavioral changes. In mouse embryonic stem cells, AUTS2 could promote or delay neuronal differentiation. Cerebral organoids, derived from an AUTS2 syndrome patient containing a pathogenic missense variant in exon 9, exhibited neocortical growth defects. Emerging technologies for analysis of human cerebral organoids will be increasingly useful for understanding mechanisms underlying AUTS2 syndrome. Questions for future research include whether AUTS2 binds RNA directly, how AUTS2 regulates neurogenesis, and how AUTS2 modulates neural circuit formation.
Keywords: AUTS2 syndrome; FBRSL1; RNA-binding protein; cerebellar hypoplasia; cerebral organoids; dentate gyrus hypoplasia; intellectual disability; microcephaly.
Copyright © 2022 Biel, Castanza, Rutherford, Fair, Chifamba, Wester, Hester and Hevner.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures