

THOR's Hammer: the Antibiotic Koreenceine Drives Gene Expression in a Model Microbial Community
- PMID: 35435700
- PMCID: PMC9239112
- DOI: 10.1128/mbio.02486-21
THOR's Hammer: the Antibiotic Koreenceine Drives Gene Expression in a Model Microbial Community
Abstract
Microbial interactions dictate the structure and function of microbiomes, but the complexity of natural communities can obscure the individual interactions. Model microbial communities constructed with genetically tractable strains known to interact in natural settings can untangle these networks and reveal underpinning mechanisms. Our model system,
Keywords: antibiotic signaling; microbial communities; rhizosphere microbes; transcriptional regulation.
Conflict of interest statement
The authors declare a conflict of interest. J.H. holds equity in Wacasa, Inc.
Figures
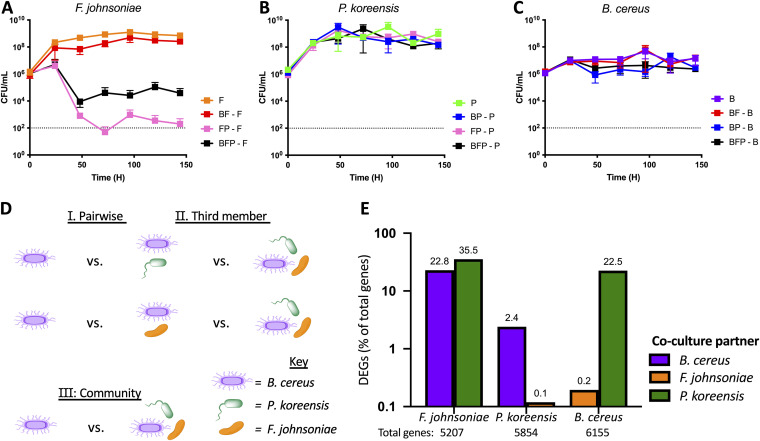
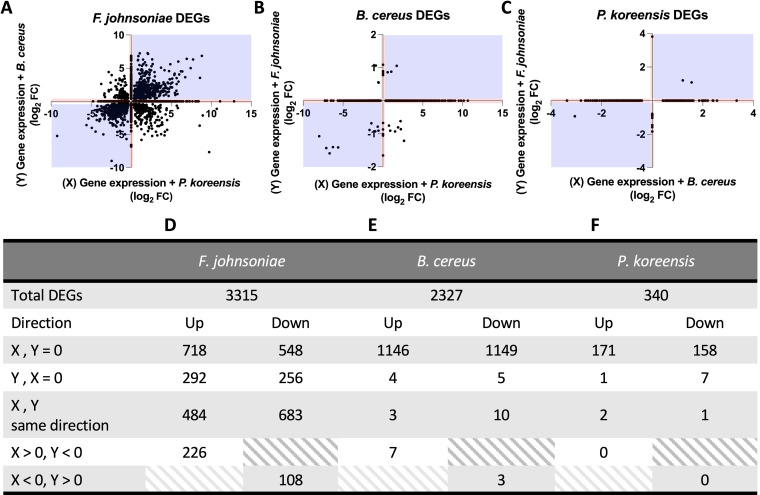
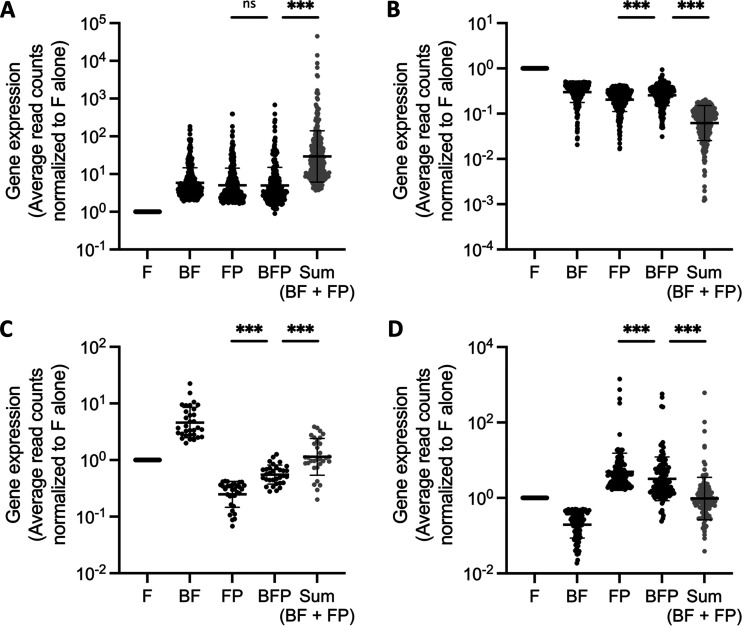
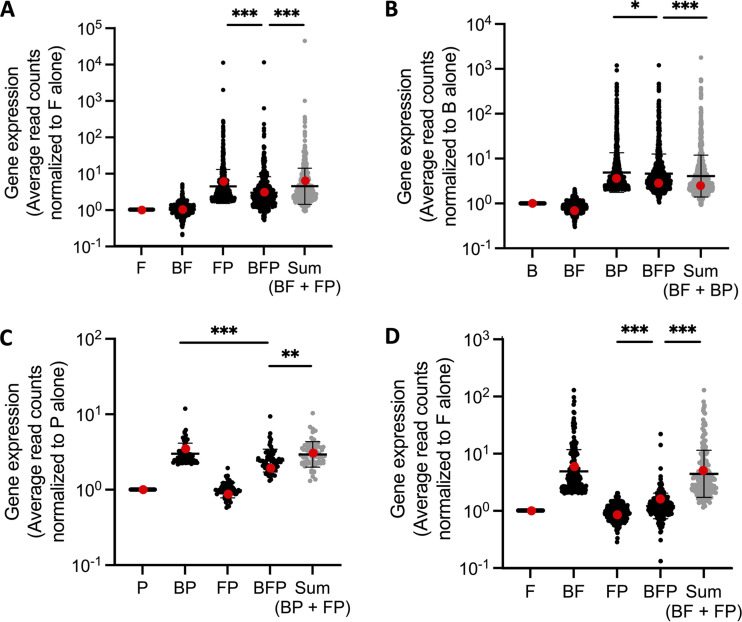
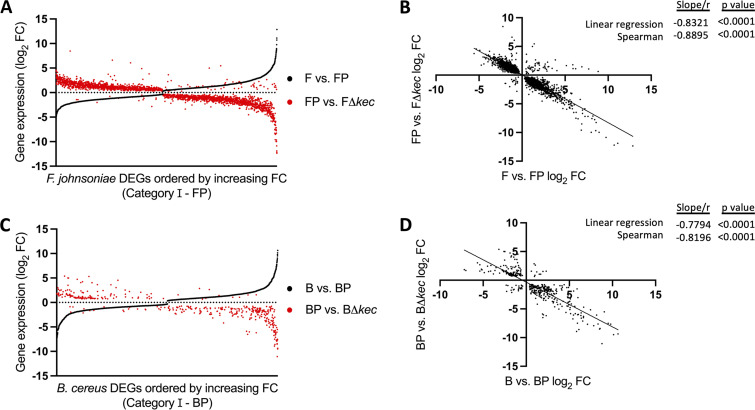
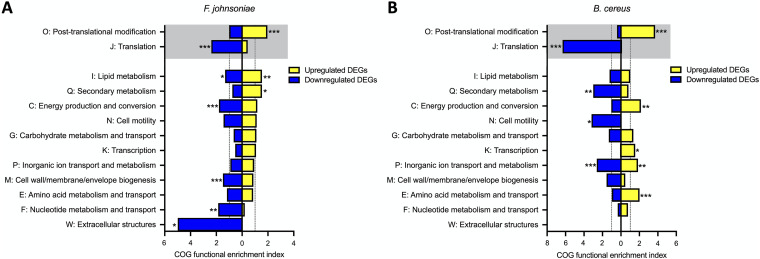
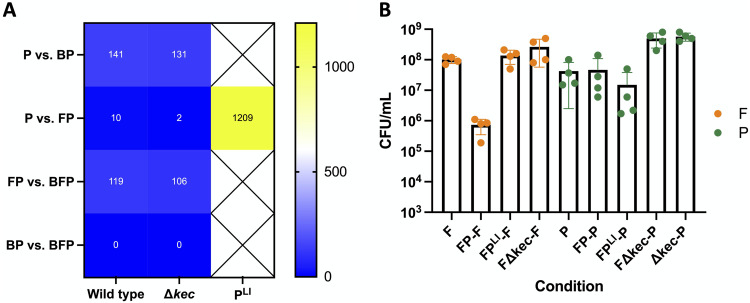
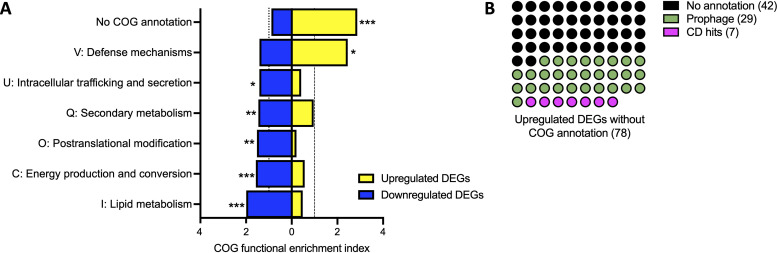
References
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
