

A proteogenomic analysis of clear cell renal cell carcinoma in a Chinese population
- PMID: 35440542
- PMCID: PMC9019091
- DOI: 10.1038/s41467-022-29577-x
A proteogenomic analysis of clear cell renal cell carcinoma in a Chinese population
Abstract
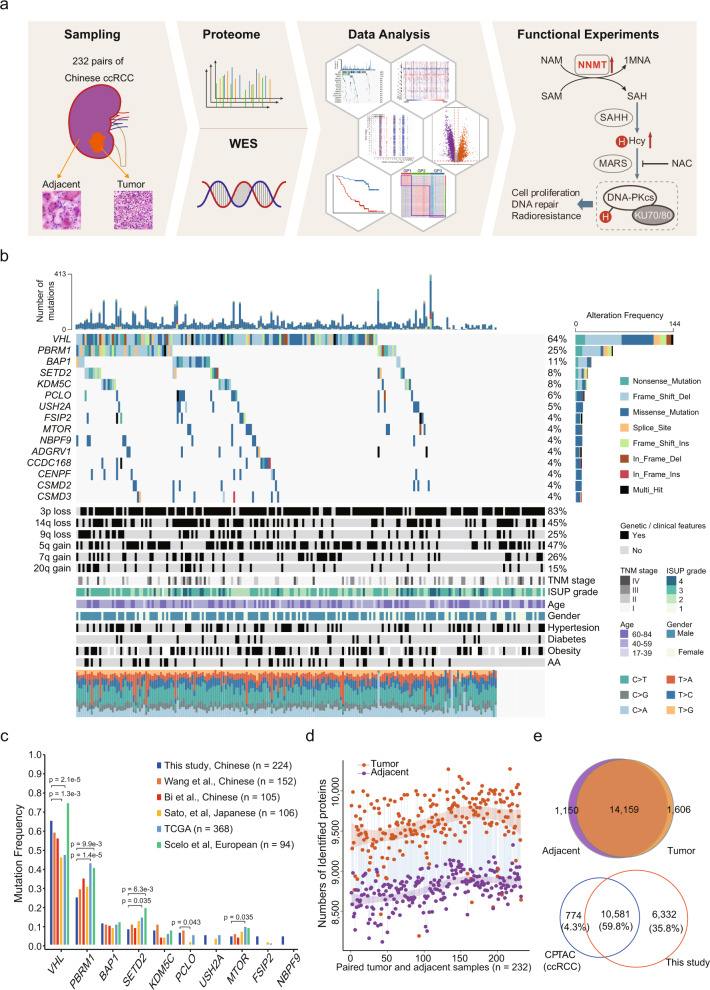
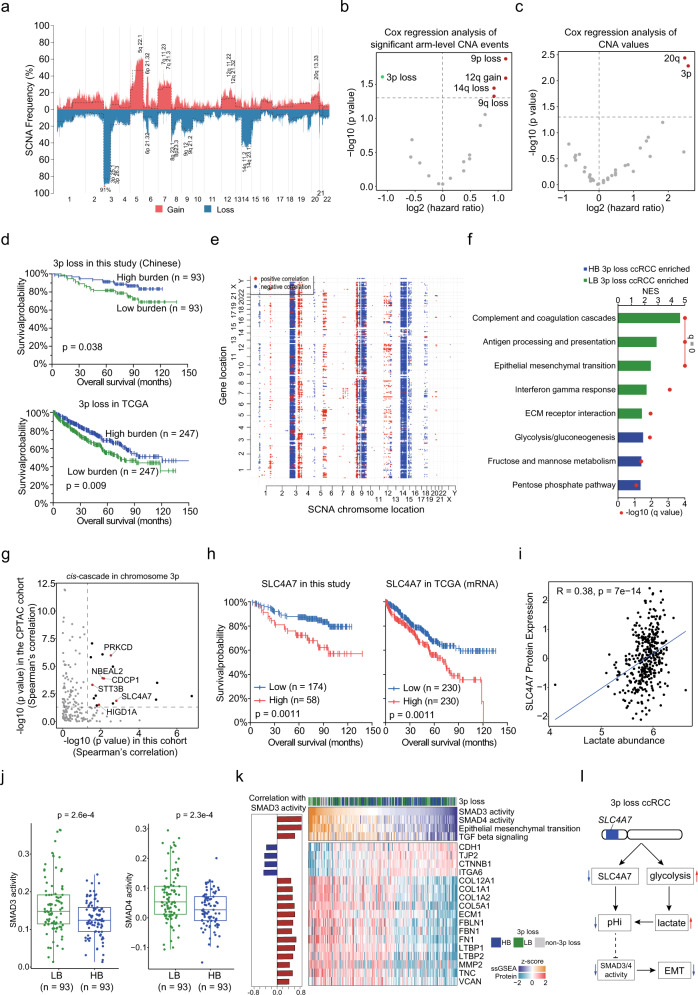
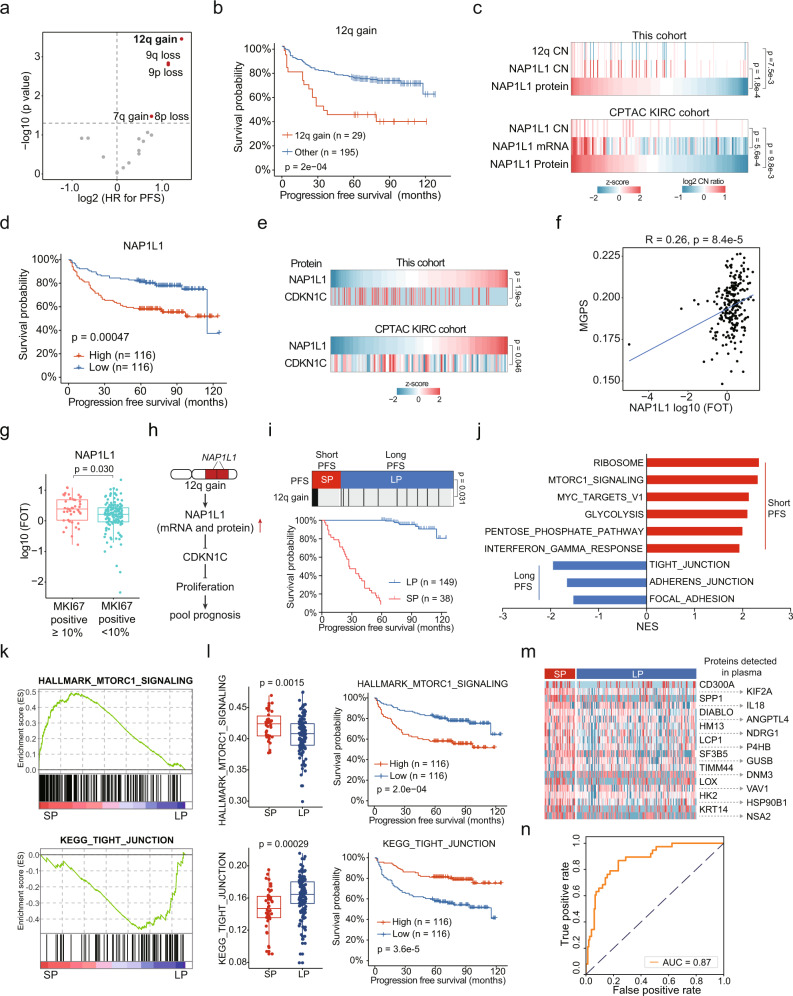
Clear cell renal cell carcinoma (ccRCC) is a common and aggressive subtype of renal cancer. Here we conduct a comprehensive proteogenomic analysis of 232 tumor and adjacent non-tumor tissue pairs from Chinese ccRCC patients. By comparing with tumor adjacent tissues, we find that ccRCC shows extensive metabolic dysregulation and an enhanced immune response. Molecular subtyping classifies ccRCC tumors into three subtypes (GP1-3), among which the most aggressive GP1 exhibits the strongest immune phenotype, increased metastasis, and metabolic imbalance, linking the multi-omics-derived phenotypes to clinical outcomes of ccRCC. Nicotinamide N-methyltransferase (NNMT), a one-carbon metabolic enzyme, is identified as a potential marker of ccRCC and a drug target for GP1. We demonstrate that NNMT induces DNA-dependent protein kinase catalytic subunit (DNA-PKcs) homocysteinylation, increases DNA repair, and promotes ccRCC tumor growth. This study provides insights into the biological underpinnings and prognosis assessment of ccRCC, revealing targetable metabolic vulnerabilities.
© 2022. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures




References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
