

Genomics, convergent neuroscience and progress in understanding autism spectrum disorder
- PMID: 35440779
- PMCID: PMC10693992
- DOI: 10.1038/s41583-022-00576-7
Genomics, convergent neuroscience and progress in understanding autism spectrum disorder
Abstract
More than a hundred genes have been identified that, when disrupted, impart large risk for autism spectrum disorder (ASD). Current knowledge about the encoded proteins - although incomplete - points to a very wide range of developmentally dynamic and diverse biological processes. Moreover, the core symptoms of ASD involve distinctly human characteristics, presenting challenges to interpreting evolutionarily distant model systems. Indeed, despite a decade of striking progress in gene discovery, an actionable understanding of pathobiology remains elusive. Increasingly, convergent neuroscience approaches have been recognized as an important complement to traditional uses of genetics to illuminate the biology of human disorders. These methods seek to identify intersection among molecular-level, cellular-level and circuit-level functions across multiple risk genes and have highlighted developing excitatory neurons in the human mid-gestational prefrontal cortex as an important pathobiological nexus in ASD. In addition, neurogenesis, chromatin modification and synaptic function have emerged as key potential mediators of genetic vulnerability. The continued expansion of foundational 'omics' data sets, the application of higher-throughput model systems and incorporating developmental trajectories and sex differences into future analyses will refine and extend these results. Ultimately, a systems-level understanding of ASD genetic risk holds promise for clarifying pathobiology and advancing therapeutics.
© 2022. Springer Nature Limited.
Conflict of interest statement
Competing interests
The authors declare no competing interests.
Figures
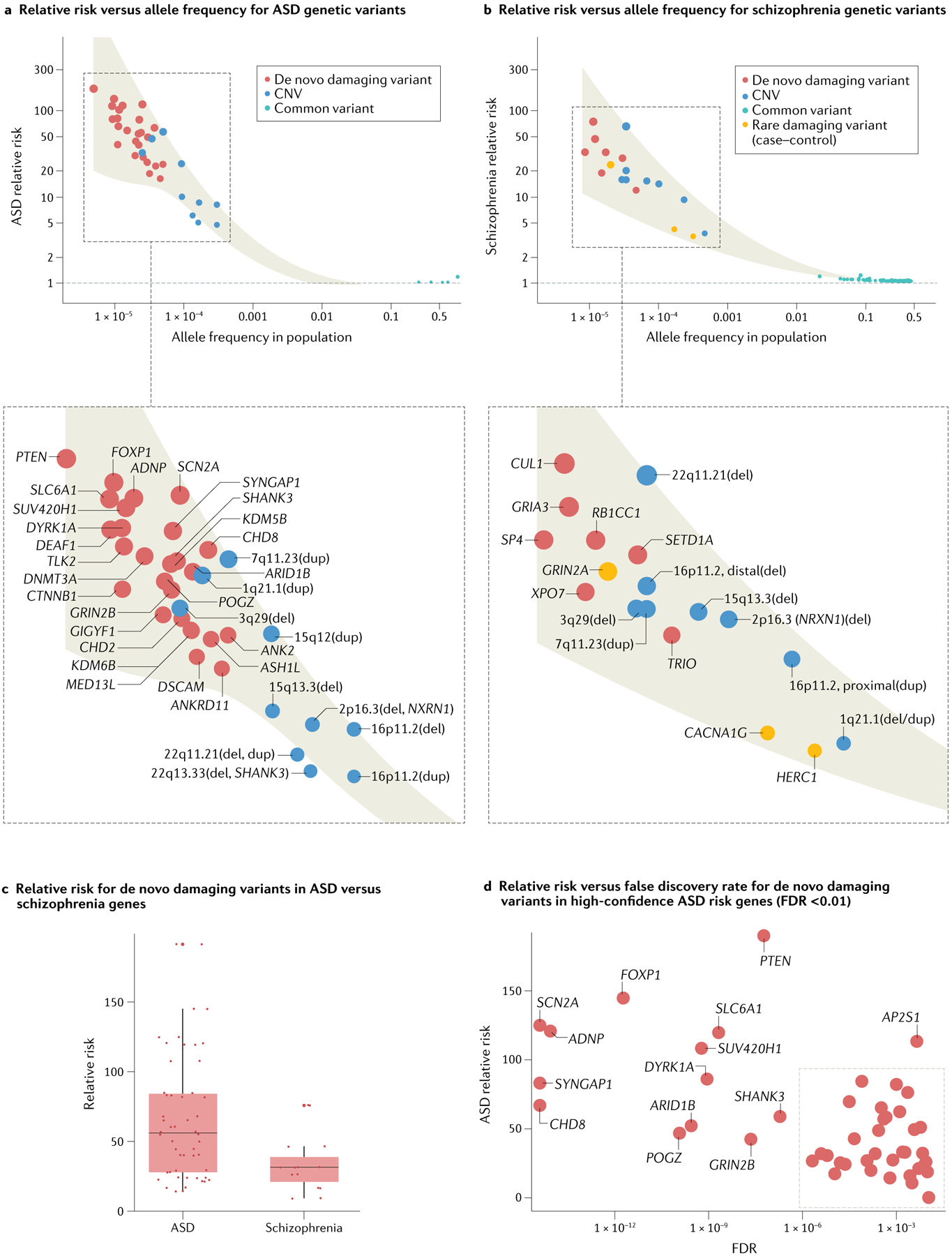
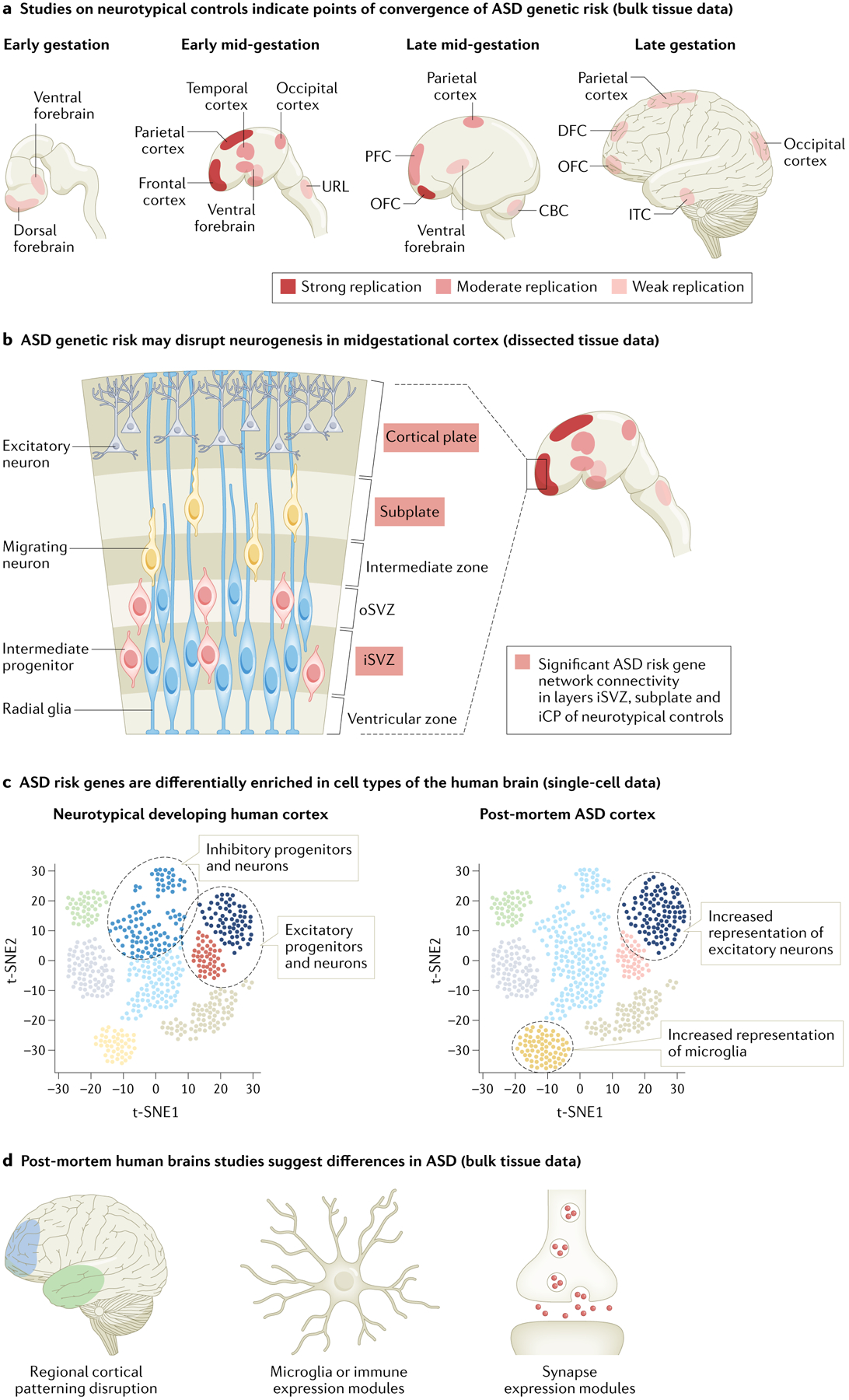
References
-
- Díaz-Caneja CM et al. A white paper on a neurodevelopmental framework for drug discovery in autism and other neurodevelopmental disorders. Eur. Neuropsychopharmacol 48, 49–88 (2021). - PubMed
-
- Folstein S & Rutter M Infantile autism: a genetic study of 21 twin pairs. J. Child. Psychol. Psychiatry 18, 297–321 (1977). - PubMed
