

A randomized, double-blind trial of triheptanoin for drug-resistant epilepsy in glucose transporter 1 deficiency syndrome
- PMID: 35441706
- PMCID: PMC9546029
- DOI: 10.1111/epi.17263
A randomized, double-blind trial of triheptanoin for drug-resistant epilepsy in glucose transporter 1 deficiency syndrome
Abstract
Objective: This study was undertaken to evaluate efficacy and long-term safety of triheptanoin in patients >1 year old, not on a ketogenic diet, with drug-resistant seizures associated with glucose transporter 1 deficiency syndrome (Glut1DS).
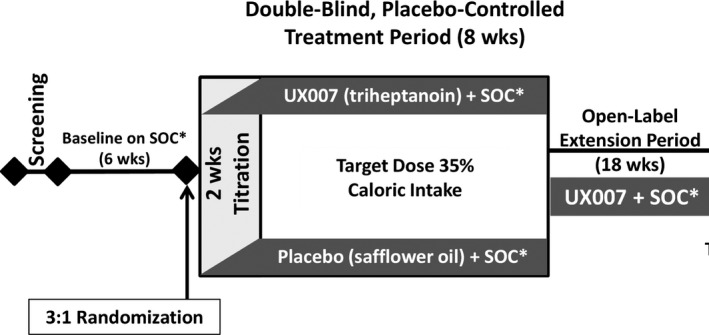
Methods: UX007G-CL201 was a randomized, double-blind, placebo-controlled trial. Following a 6-week baseline period, eligible patients were randomized 3:1 to triheptanoin or placebo. Dosing was titrated to 35% of total daily calories over 2 weeks. After an 8-week placebo-controlled period, all patients received open-label triheptanoin through Week 52.
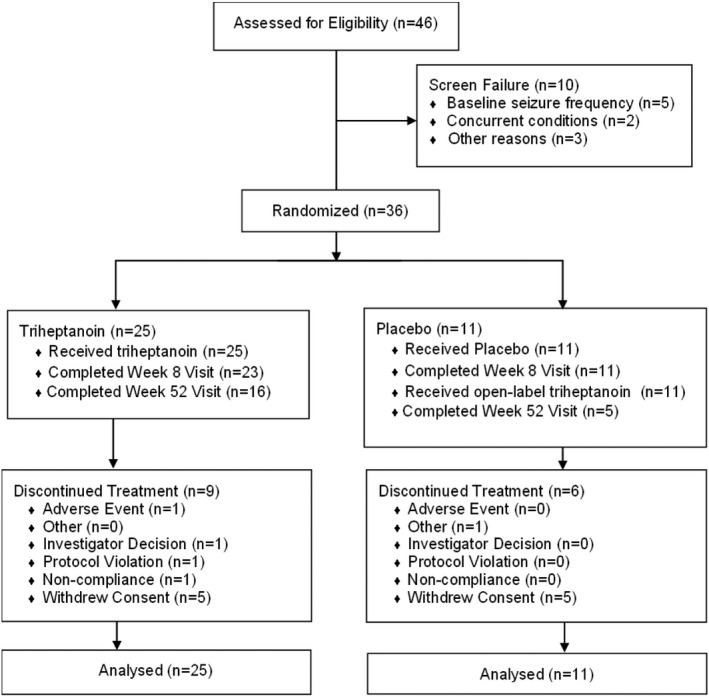
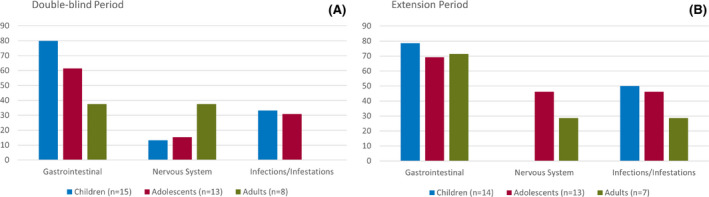
Results: The study included 36 patients (15 children, 13 adolescents, eight adults). A median 12.6% reduction in overall seizure frequency was observed in the triheptanoin arm relative to baseline, and a 13.5% difference was observed relative to placebo (p = .58). In patients with absence seizures only (n = 9), a median 62.2% reduction in seizure frequency was observed in the triheptanoin arm relative to baseline. Only one patient with absence seizures only was present in the control group, preventing comparison. No statistically significant differences in seizure frequency were observed. Common treatment-emergent adverse events included diarrhea, vomiting, abdominal pain, and nausea, mostly mild or moderate in severity. No serious adverse events were considered to be treatment related. One patient discontinued due to status epilepticus.
Significance: Triheptanoin did not significantly reduce seizure frequency in patients with Glut1DS not on the ketogenic diet. Treatment was associated with mild to moderate gastrointestinal treatment-related events; most resolved following dose reduction or interruption and/or medication for treatment. Triheptanoin was not associated with any long-term safety concerns when administered at dose levels up to 35% of total daily caloric intake for up to 1 year.
Trial registration: ClinicalTrials.gov NCT01993186.
Keywords: diet treatment; drug resistance; epilepsy; glucose transporter 1 deficiency syndrome; triheptanoin.
© 2022 The Authors. Epilepsia published by Wiley Periodicals LLC on behalf of International League Against Epilepsy.
Conflict of interest statement
P.S. has received fees from Ultragenyx Pharmaceutical Inc, Zogenix, BioMarin, PTC Therapeutics, GW Pharma, and Neuraxpharm, and research grants from GW Pharma, PTC Therapeutics, Enecta, and Kolfarma. He has been an investigator for clinical trials for Ultragenyx Pharmaceutical Inc and Zogenix. He has served on a scientific advisory board for the Italian Medicines Agency; has received honoraria from GW Pharma, Kolfarma, and Eisai; and has received research support from the Italian Ministry of Health and San Paolo Foundation. S.A. has served as a consultant or received honoraria for lectures from Biocodex, BioMarin, Eisai, GW Pharma, Neuraxpharm, Nutricia, UCB Pharma, Ultragenyx Pharmaceutical Inc, and Zogenix. He has been an investigator for clinical trials for Eisai, UCB Pharma, Ultragenyx Pharmaceutical Inc, and Zogenix. I.E.S. has served on scientific advisory boards for UCB, Eisai, GlaxoSmithKline, BioMarin, Nutricia, Rogcon, Chiesi, Encoded Therapeutics, Knopp Biosciences, and Xenon Pharmaceuticals; has received speaker honoraria from GlaxoSmithKline, UCB, BioMarin, Biocodex, Chiesi, LivaNova, and Eisai; has received funding for travel from UCB, Biocodex, GlaxoSmithKline, BioMarin, and Eisai; has served as an investigator for Zogenix, Zynerba, Ultragenyx Pharmaceutical Inc, GW Pharma, UCB, Eisai, Xenon Pharmaceuticals, Anavex Life Sciences, Ovid Therapeutics, Epygenix Therapeutics, Encoded Therapeutics, and Marinus; and has consulted for Zynerba Pharmaceuticals, Atheneum Partners, Ovid Therapeutics, Care Beyond Diagnosis, Epilepsy Consortium, and UCB. She may accrue future revenue on pending patent WO61/010176 (filed 2008; Therapeutic Compound); has a patent for
Figures
References
-
- Pearson TS, Akman C, Hinton VJ, Engelstad K, De Vivo DC. Phenotypic spectrum of glucose transporter type 1 deficiency syndrome (Glut1 DS). Curr Neurol Neurosci Rep. 2013;13(4):342. - PubMed
-
- De Vivo DC, Trifiletti RR, Jacobson RI, Ronen GM, Behmand RA, Harik SI. Defective glucose transport across the blood‐brain barrier as a cause of persistent hypoglycorrhachia, seizures, and developmental delay. N Engl J Med. 1991;325(10):703–9. - PubMed
-
- Koch H, Weber YG. The glucose transporter type 1 (Glut1) syndromes. Epilepsy Behav. 2019;91:90–3. - PubMed
-
- Coman DJ, Sinclair KG, Burke CJ, Appleton DB, Pelekanos JT, O'Neil CM, et al. Seizures, ataxia, developmental delay and the general paediatrician: glucose transporter 1 deficiency syndrome. J Paediatr Child Health. 2006;42(5):263–7. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Associated data
LinkOut - more resources
Full Text Sources
Medical
