

Acute Leukemia Classification Using Transcriptional Profiles From Low-Cost Nanopore mRNA Sequencing
- PMID: 35442720
- PMCID: PMC9200386
- DOI: 10.1200/PO.21.00326
Acute Leukemia Classification Using Transcriptional Profiles From Low-Cost Nanopore mRNA Sequencing
Abstract
Purpose: Most cases of pediatric acute leukemia occur in low- and middle-income countries, where health centers lack the tools required for accurate diagnosis and disease classification. Recent research shows the robustness of using unbiased short-read RNA sequencing to classify genomic subtypes of acute leukemia. Compared with short-read sequencing, nanopore sequencing has low capital and consumable costs, making it suitable for use in locations with limited health infrastructure.
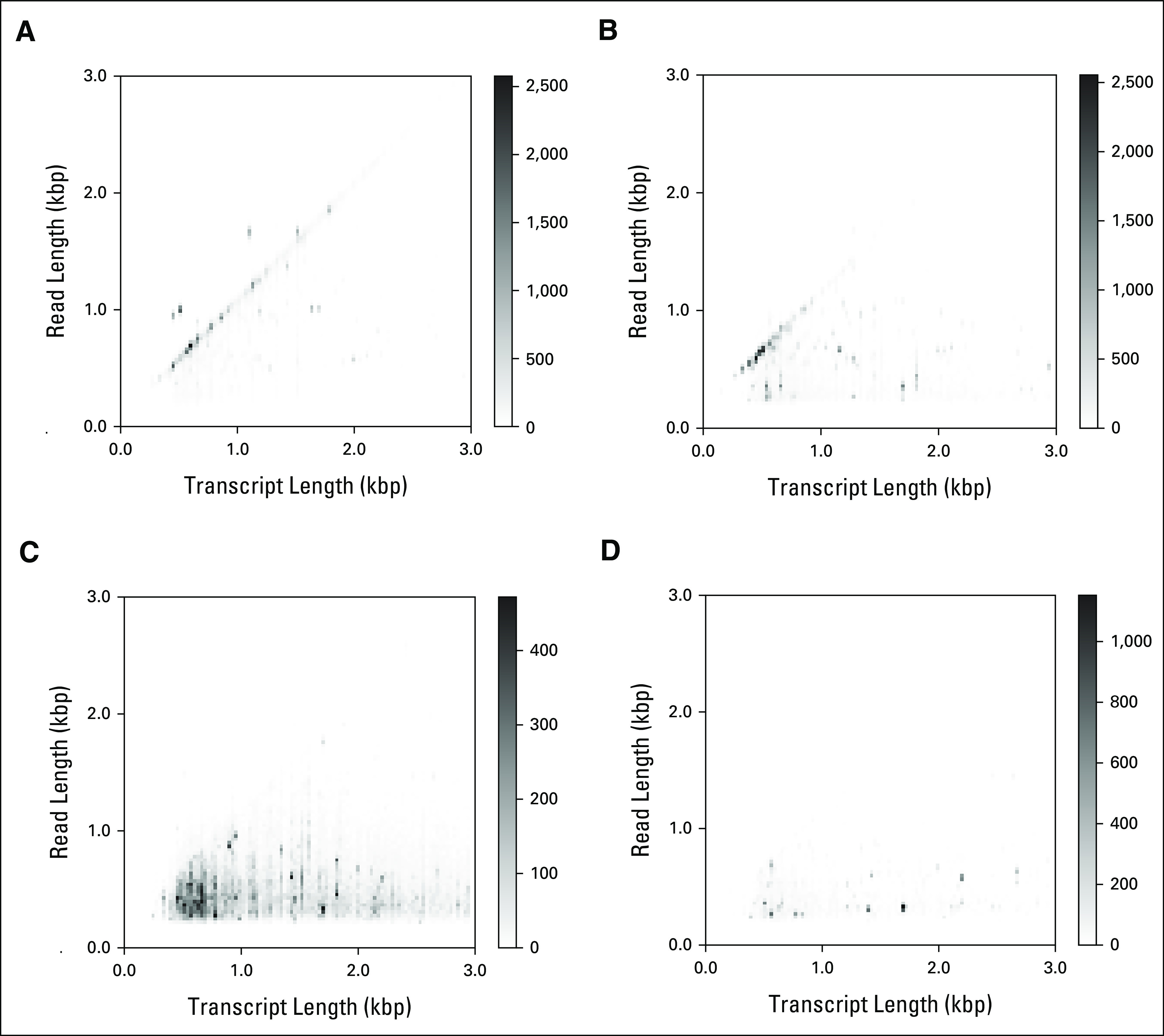
Materials and methods: We show the feasibility of nanopore mRNA sequencing on 134 cryopreserved acute leukemia specimens (26 acute myeloid leukemia [AML], 73 B-lineage acute lymphoblastic leukemia [B-ALL], 34 T-lineage acute lymphoblastic leukemia, and one acute undifferentiated leukemia). Using multiple library preparation approaches, we generated long-read transcripts for each sample. We developed a novel composite classification approach to predict acute leukemia lineage and major B-ALL and AML molecular subtypes directly from gene expression profiles.
Results: We demonstrate accurate classification of acute leukemia samples into AML, B-ALL, or T-lineage acute lymphoblastic leukemia (96.2% of cases are classifiable with a probability of > 0.8, with 100% accuracy) and further classification into clinically actionable genomic subtypes using shallow RNA nanopore sequencing, with 96.2% accuracy for major AML subtypes and 94.1% accuracy for major B-lineage acute lymphoblastic leukemia subtypes.
Conclusion: Transcriptional profiling of acute leukemia samples using nanopore technology for diagnostic classification is feasible and accurate, which has the potential to improve the accuracy of cancer diagnosis in low-resource settings.
Conflict of interest statement
Figures


References
-
- Bray F, Soerjomataram I: The changing global burden of cancer: Transitions in human development and implications for cancer prevention and control, in Gelband H, Jha P, Sankaranarayanan R, et al. (eds): Cancer: Disease Control Priorities, Volume 3 (ed 2). Washington (DC), The International Bank for Reconstruction and Development/The World Bank, 2015 - PubMed
-
- Bhakta N, Force LM, Allemani C, et al. : Childhood cancer burden: A review of global estimates. Lancet Oncol 20:e42-e53, 2019 - PubMed
-
- Bray F, Ferlay J, Soerjomataram I, et al. : Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 68:394-424, 2018 - PubMed
-
- Ward ZJ, Yeh JM, Bhakta N, et al. : Estimating the total incidence of global childhood cancer: A simulation-based analysis. Lancet Oncol 20:483-493, 2019 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
