

All2: A tool for selecting mosaic mutations from comprehensive multi-cell comparisons
- PMID: 35442945
- PMCID: PMC9060341
- DOI: 10.1371/journal.pcbi.1009487
All2: A tool for selecting mosaic mutations from comprehensive multi-cell comparisons
Erratum in
-
Correction: All2: A tool for selecting mosaic mutations from comprehensive multi-cell comparisons.PLoS Comput Biol. 2022 Nov 15;18(11):e1010703. doi: 10.1371/journal.pcbi.1010703. eCollection 2022 Nov. PLoS Comput Biol. 2022. PMID: 36378632 Free PMC article.
Abstract
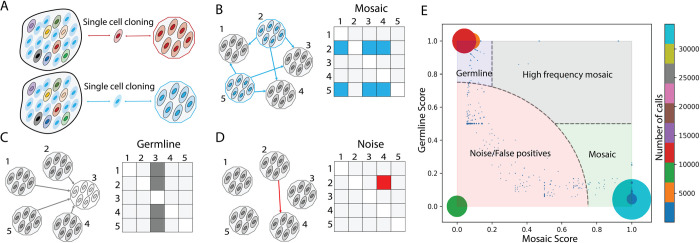
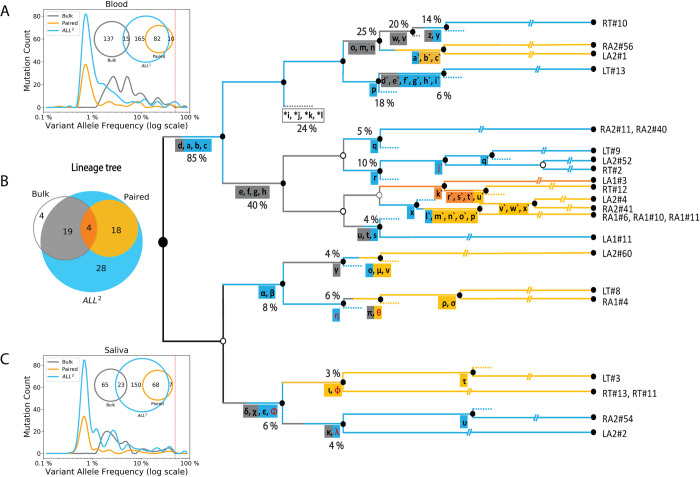
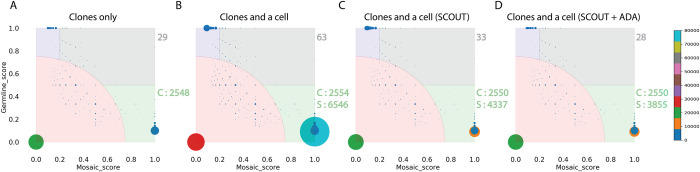
Accurate discovery of somatic mutations in a cell is a challenge that partially lays in immaturity of dedicated analytical approaches. Approaches comparing a cell's genome to a control bulk sample miss common mutations, while approaches to find such mutations from bulk suffer from low sensitivity. We developed a tool, All2, which enables accurate filtering of mutations in a cell without the need for data from bulk(s). It is based on pair-wise comparisons of all cells to each other where every call for base pair substitution and indel is classified as either a germline variant, mosaic mutation, or false positive. As All2 allows for considering dropped-out regions, it is applicable to whole genome and exome analysis of cloned and amplified cells. By applying the approach to a variety of available data, we showed that its application reduces false positives, enables sensitive discovery of high frequency mutations, and is indispensable for conducting high resolution cell lineage tracing.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
