

The Evolution and Biology of SARS-CoV-2 Variants
- PMID: 35444005
- PMCID: PMC9159258
- DOI: 10.1101/cshperspect.a041390
The Evolution and Biology of SARS-CoV-2 Variants
Abstract
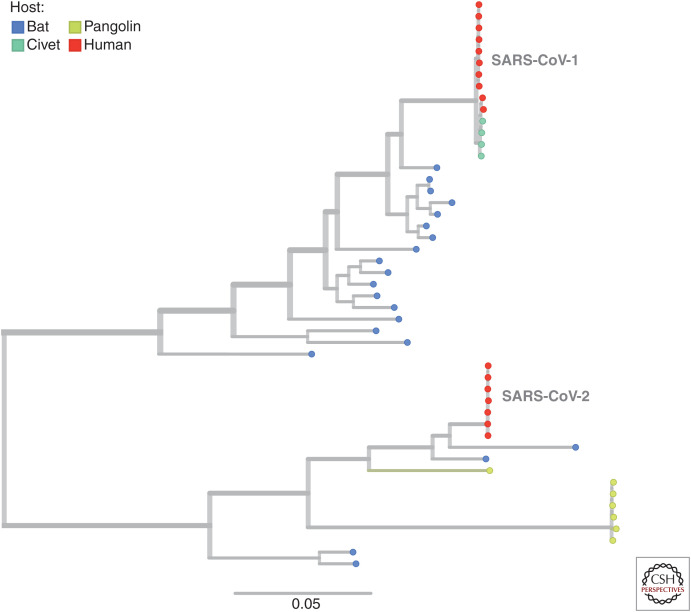
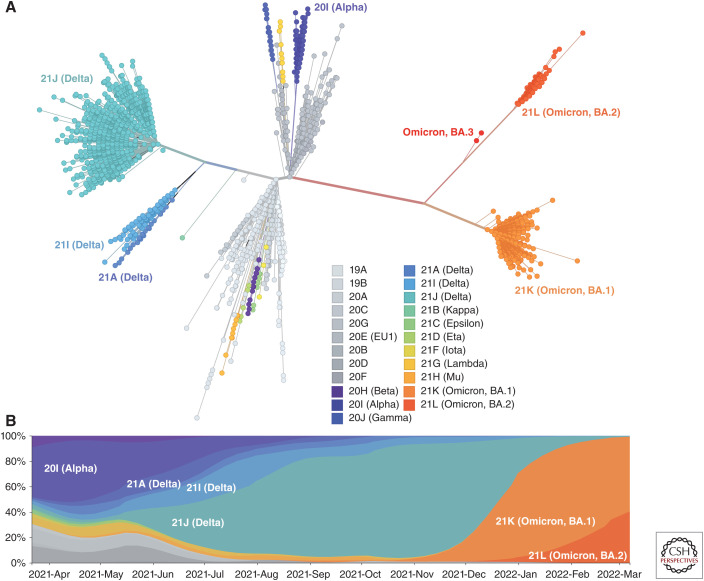
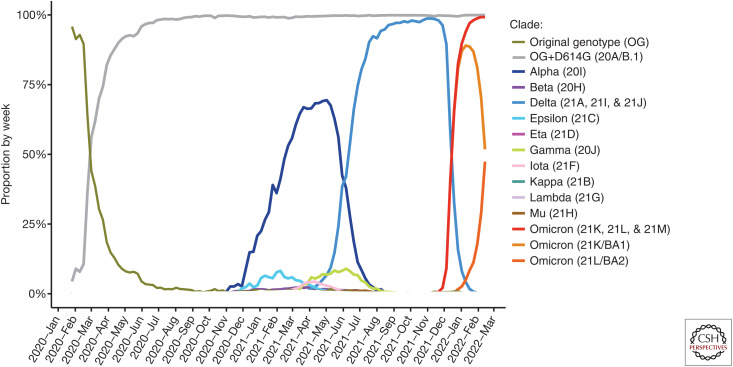
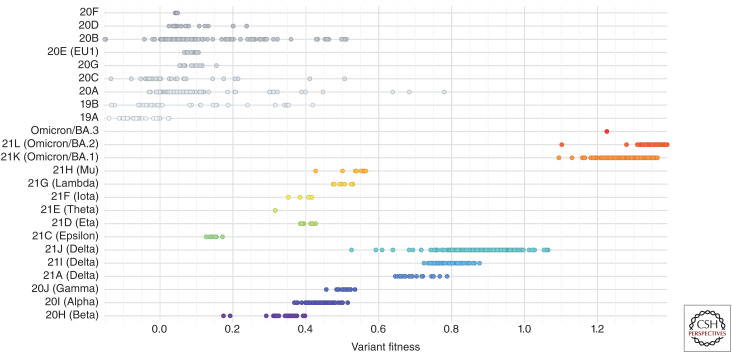
Our understanding of the still unfolding severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) pandemic would have been extremely limited without the study of the genetics and evolution of this new human coronavirus. Large-scale genome-sequencing efforts have provided close to real-time tracking of the global spread and diversification of SARS-CoV-2 since its entry into the human population in late 2019. These data have underpinned analysis of its origins, epidemiology, and adaptations to the human population: principally immune evasion and increasing transmissibility. SARS-CoV-2, despite being a new human pathogen, was highly capable of human-to-human transmission. During its rapid spread in humans, SARS-CoV-2 has evolved independent new forms, the so-called "variants of concern," that are better optimized for human-to-human transmission. The most important adaptation of the bat coronavirus progenitor of both SARS-CoV-1 and SARS-CoV-2 for human infection (and other mammals) is the use of the angiotensin-converting enzyme 2 (ACE2) receptor. Relaxed structural constraints provide plasticity to SARS-related coronavirus spike protein permitting it to accommodate significant amino acid replacements of antigenic consequence without compromising the ability to bind to ACE2. Although the bulk of research has justifiably concentrated on the viral spike protein as the main determinant of antigenic evolution and changes in transmissibility, there is accumulating evidence for the contribution of other regions of the viral proteome to virus-host interaction. Whereas levels of community transmission of recombinants compromising genetically distinct variants are at present low, when divergent variants cocirculate, recombination between SARS-CoV-2 clades is being detected, increasing the risk that viruses with new properties emerge. Applying computational and machine learning methods to genome sequence data sets to generate experimentally verifiable predictions will serve as an early warning system for novel variant surveillance and will be important in future vaccine planning. Omicron, the latest SARS-CoV-2 variant of concern, has focused attention on step change antigenic events, "shift," as opposed to incremental "drift" changes in antigenicity. Both an increase in transmissibility and antigenic shift in Omicron led to it readily causing infections in the fully vaccinated and/or previously infected. Omicron's virulence, while reduced relative to the variant of concern it replaced, Delta, is very much premised on the past immune exposure of individuals with a clear signal that boosted vaccination protects from severe disease. Currently, SARS-CoV-2 has proven itself to be a dangerous new human respiratory pathogen with an unpredictable evolutionary capacity, leading to a risk of future variants too great not to ensure all regions of the world are screened by viral genome sequencing, protected through available and affordable vaccines, and have non-punitive strategies in place for detecting and responding to novel variants of concern.
Copyright © 2022 Cold Spring Harbor Laboratory Press; all rights reserved.
Figures
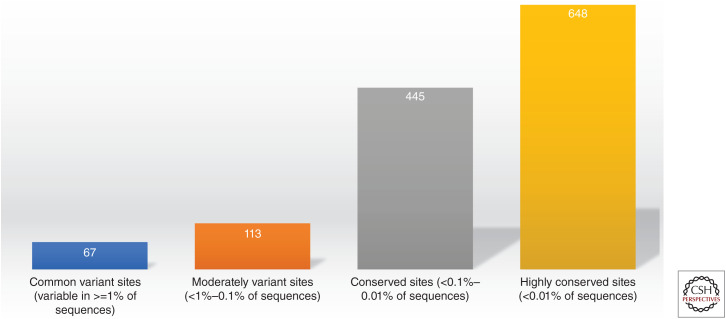
References
-
- Bedford T, Hodcroft EB, Neher RA. 2021. Updated Nextstrain SARS-CoV-2 clade naming strategy. Nextstrain Blog, January 6, 2021. https://nextstrain.org/blog/2021-01-06-updated-SARS-CoV-2-clade-naming [Accessed February 14, 2022].
-
- Bentley EG, Kirby A, Sharma P, Kipar A, Mega DF, Bramwell C, Penrice-Randal R, Prince T, Brown JC, Zhou J, et al. 2021. SARS-CoV-2 Omicron-B.1.1.529 variant leads to less severe disease than Pango B and Delta variants strains in a mouse model of severe COVID-19. bioRxiv 10.1101/2021.12.26.474085 - DOI
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous