Somatic mosaicism reveals clonal distributions of neocortical development
- PMID: 35444276
- PMCID: PMC9436791
- DOI: 10.1038/s41586-022-04602-7
Somatic mosaicism reveals clonal distributions of neocortical development
Abstract
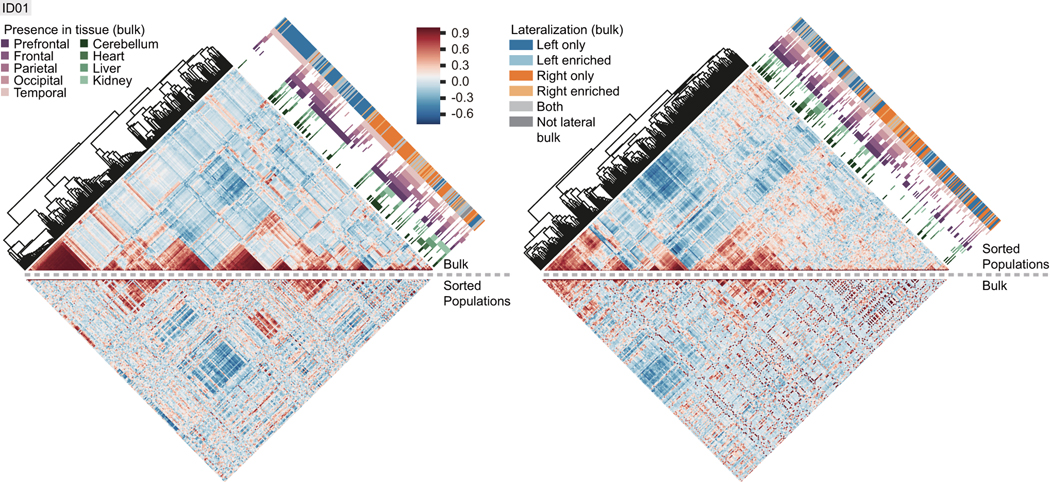
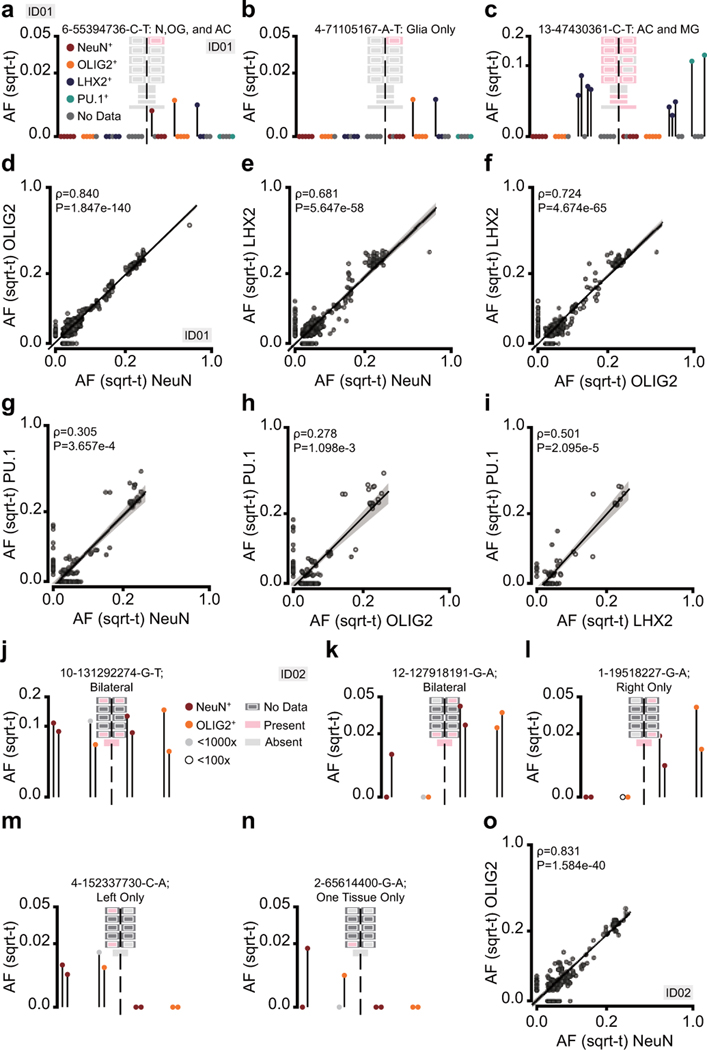
The structure of the human neocortex underlies species-specific traits and reflects intricate developmental programs. Here we sought to reconstruct processes that occur during early development by sampling adult human tissues. We analysed neocortical clones in a post-mortem human brain through a comprehensive assessment of brain somatic mosaicism, acting as neutral lineage recorders1,2. We combined the sampling of 25 distinct anatomic locations with deep whole-genome sequencing in a neurotypical deceased individual and confirmed results with 5 samples collected from each of three additional donors. We identified 259 bona fide mosaic variants from the index case, then deconvolved distinct geographical, cell-type and clade organizations across the brain and other organs. We found that clones derived after the accumulation of 90-200 progenitors in the cerebral cortex tended to respect the midline axis, well before the anterior-posterior or ventral-dorsal axes, representing a secondary hierarchy following the overall patterning of forebrain and hindbrain domains. Clones across neocortically derived cells were consistent with a dual origin from both dorsal and ventral cellular populations, similar to rodents, whereas the microglia lineage appeared distinct from other resident brain cells. Our data provide a comprehensive analysis of brain somatic mosaicism across the neocortex and demonstrate cellular origins and progenitor distribution patterns within the human brain.
© 2022. The Author(s), under exclusive licence to Springer Nature Limited.
Figures