

CCNE1 amplification is synthetic lethal with PKMYT1 kinase inhibition
- PMID: 35444283
- PMCID: PMC9046089
- DOI: 10.1038/s41586-022-04638-9
CCNE1 amplification is synthetic lethal with PKMYT1 kinase inhibition
Abstract
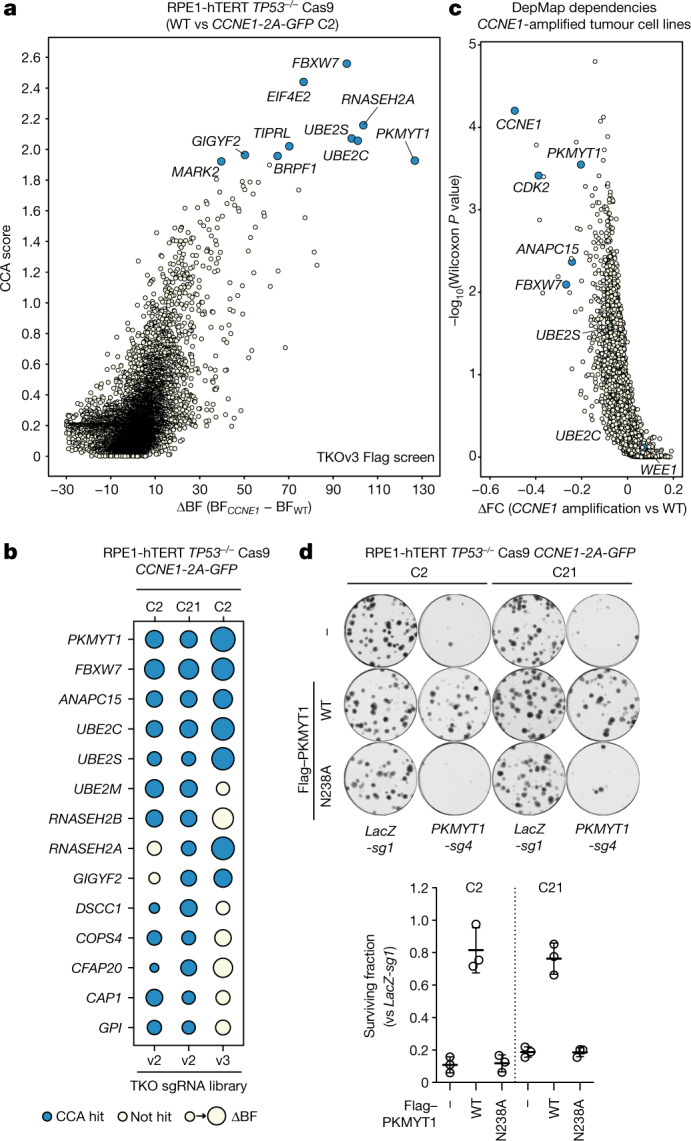
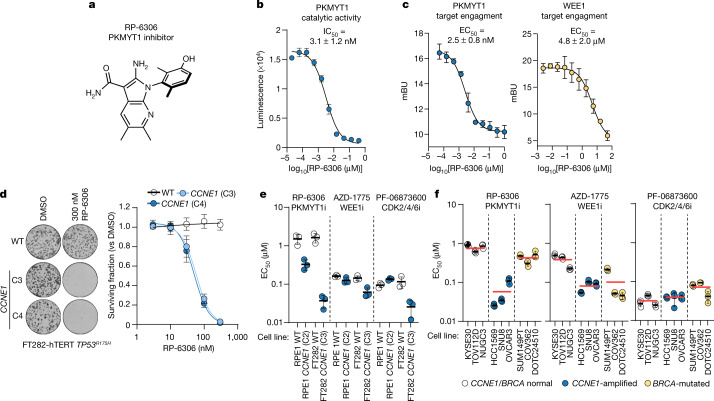
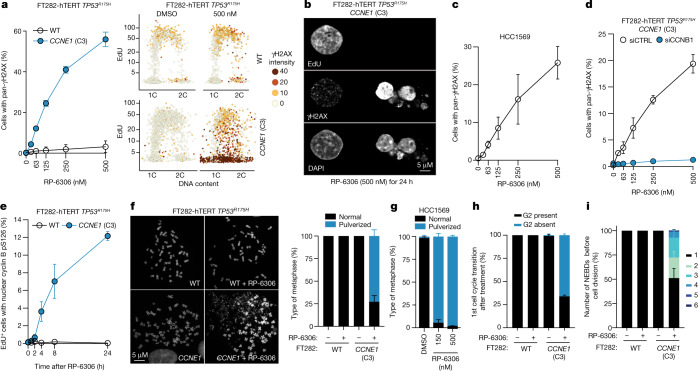
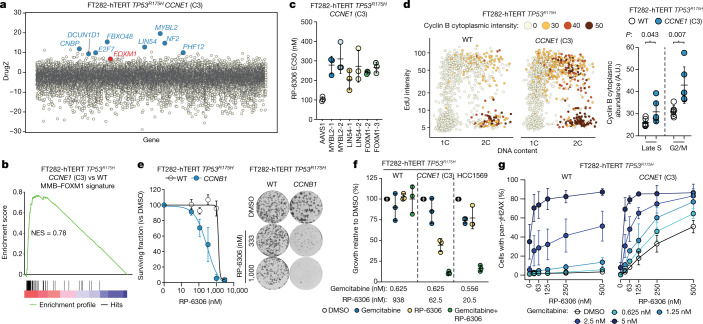
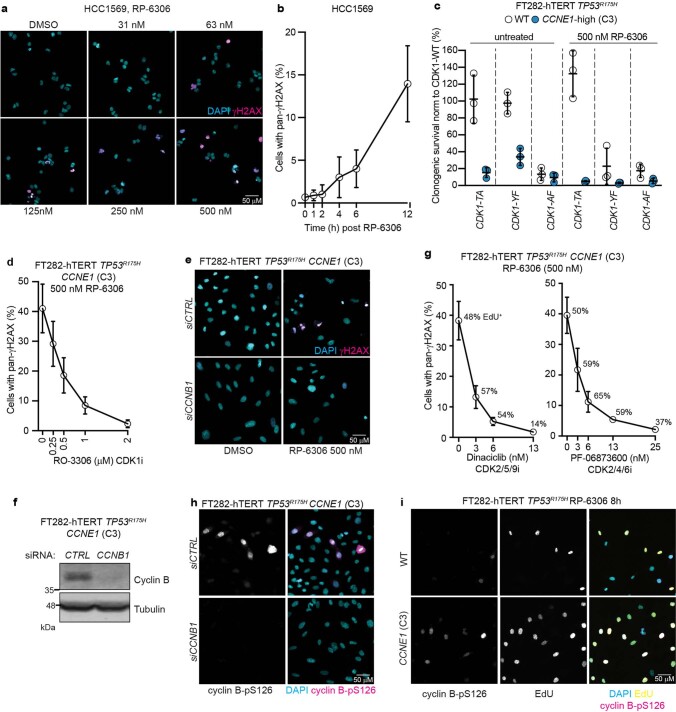
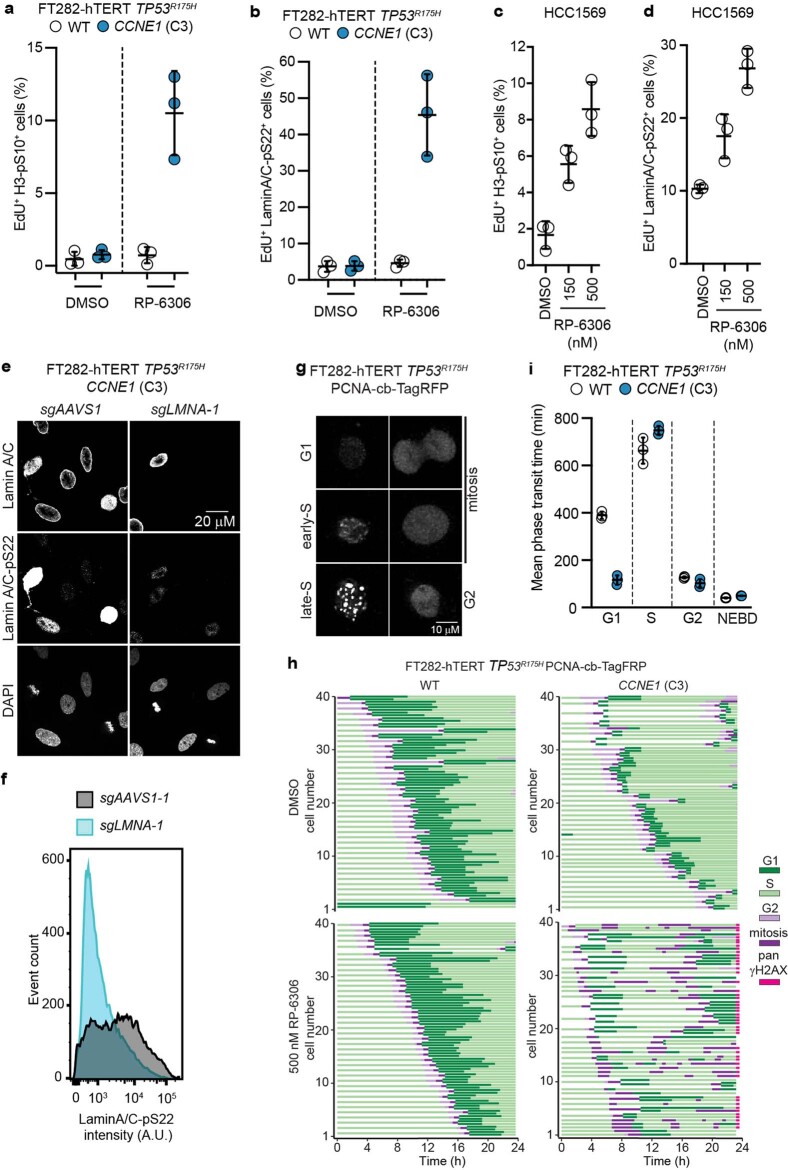
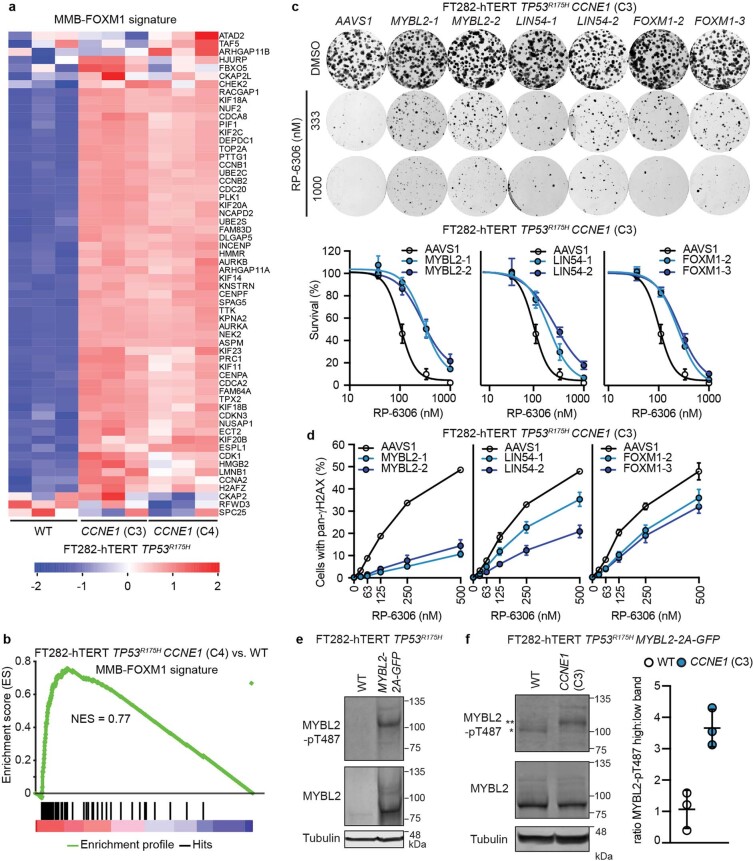
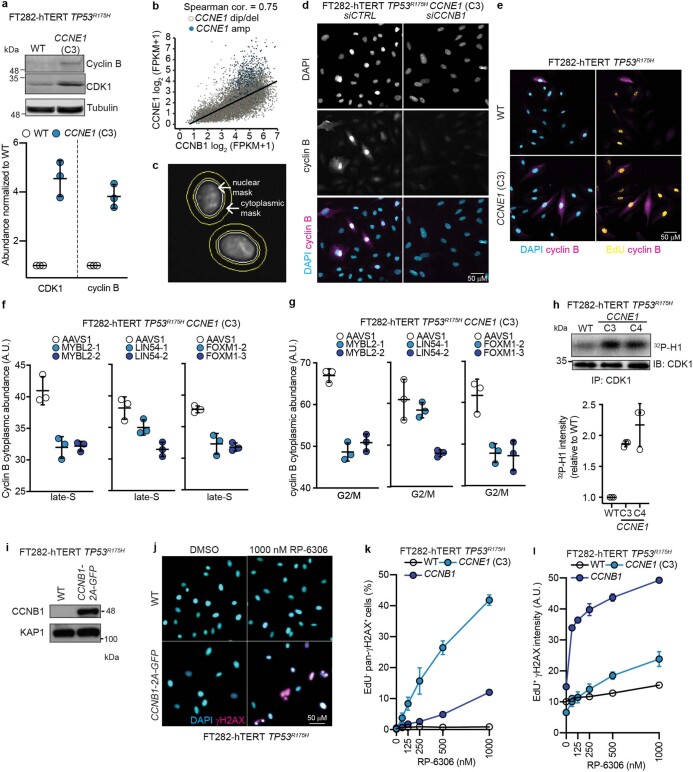
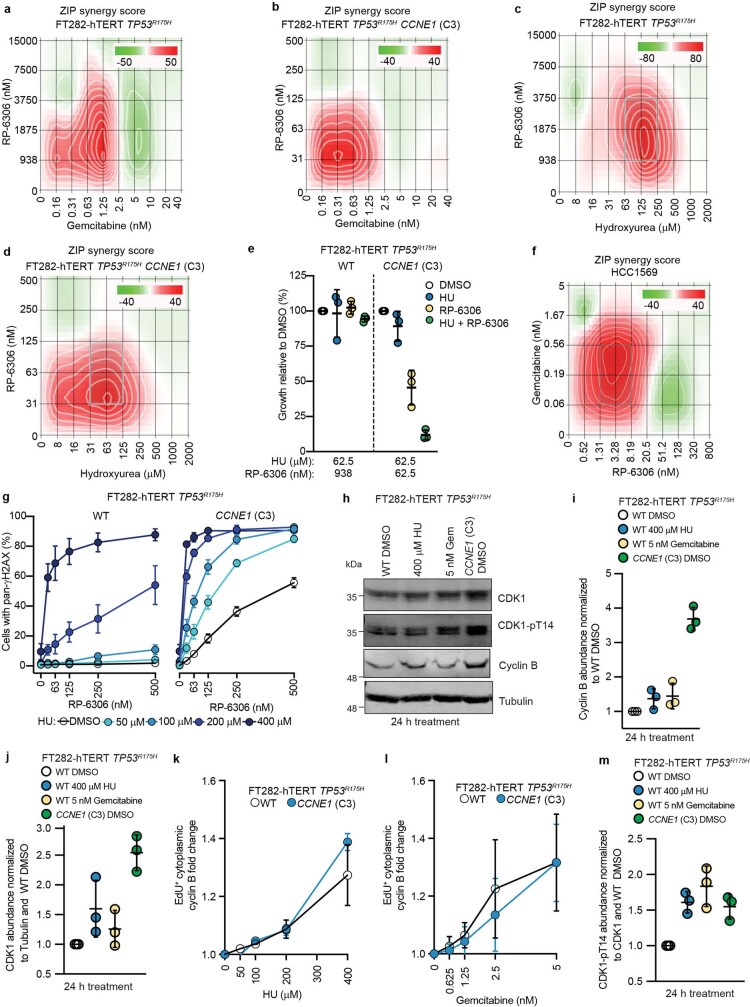
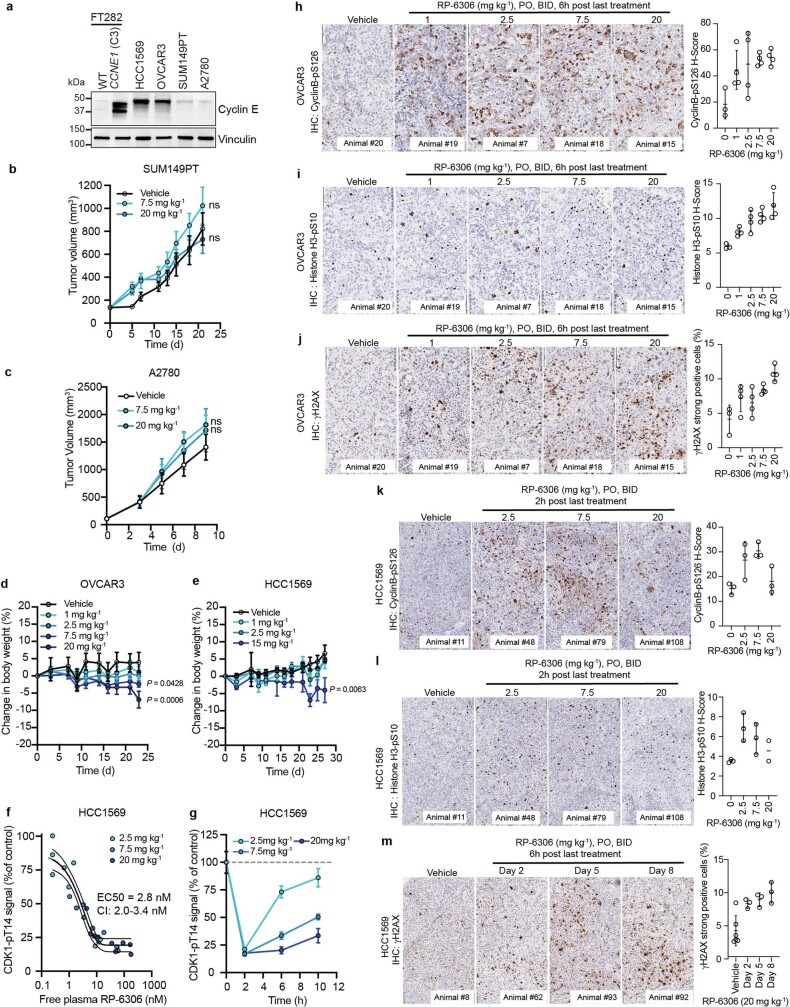
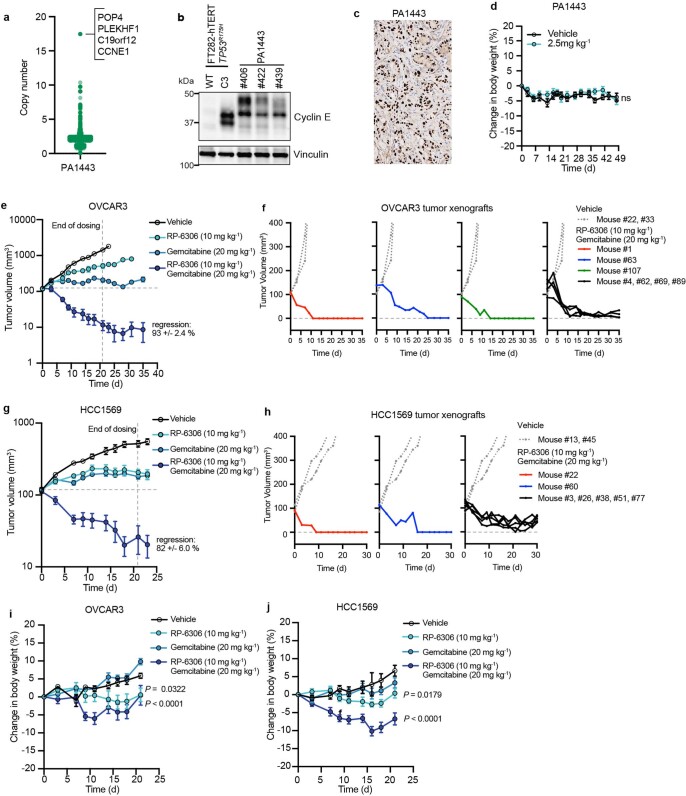
Amplification of the CCNE1 locus on chromosome 19q12 is prevalent in multiple tumour types, particularly in high-grade serous ovarian cancer, uterine tumours and gastro-oesophageal cancers, where high cyclin E levels are associated with genome instability, whole-genome doubling and resistance to cytotoxic and targeted therapies1-4. To uncover therapeutic targets for tumours with CCNE1 amplification, we undertook genome-scale CRISPR-Cas9-based synthetic lethality screens in cellular models of CCNE1 amplification. Here we report that increasing CCNE1 dosage engenders a vulnerability to the inhibition of the PKMYT1 kinase, a negative regulator of CDK1. To inhibit PKMYT1, we developed RP-6306, an orally bioavailable and selective inhibitor that shows single-agent activity and durable tumour regressions when combined with gemcitabine in models of CCNE1 amplification. RP-6306 treatment causes unscheduled activation of CDK1 selectively in CCNE1-overexpressing cells, promoting early mitosis in cells undergoing DNA synthesis. CCNE1 overexpression disrupts CDK1 homeostasis at least in part through an early activation of the MMB-FOXM1 mitotic transcriptional program. We conclude that PKMYT1 inhibition is a promising therapeutic strategy for CCNE1-amplified cancers.
© 2022. The Author(s).
Conflict of interest statement
D.D. is a shareholder, advisor and received research funding from Repare Therapeutics. D.G., J.T.F.Y., J.F., A.A.-Q., C.B., N.M.D., R.P., A.R., R.S., J.S., A.V., P.S.B., A.B.F., J.B., J.D., E.D., S.F., T.G.d.R., M.-E.L., B.L., V.B., Y.M., O.N., S.Y.Y., S.J.M., M.Z. and C.G.M. are, and G.M., H.A., C.F.-D. and A.S. were, employees of Repare Therapeutics. J.S., B.L., E.D. and R.P. have filed a patent application claiming the discovery of PKMYT1 inhibitors (WO/2021/195781) and D.D., J.T.F.Y., J.S., B.L., E.D., R.P., A.V. and J.F. have filed a patent application claiming methods of use of PKMYT1 inhibitors (WO/2021/195782). N.C., H.M., R.K.S. and J.T. declare no competing interest.
Figures
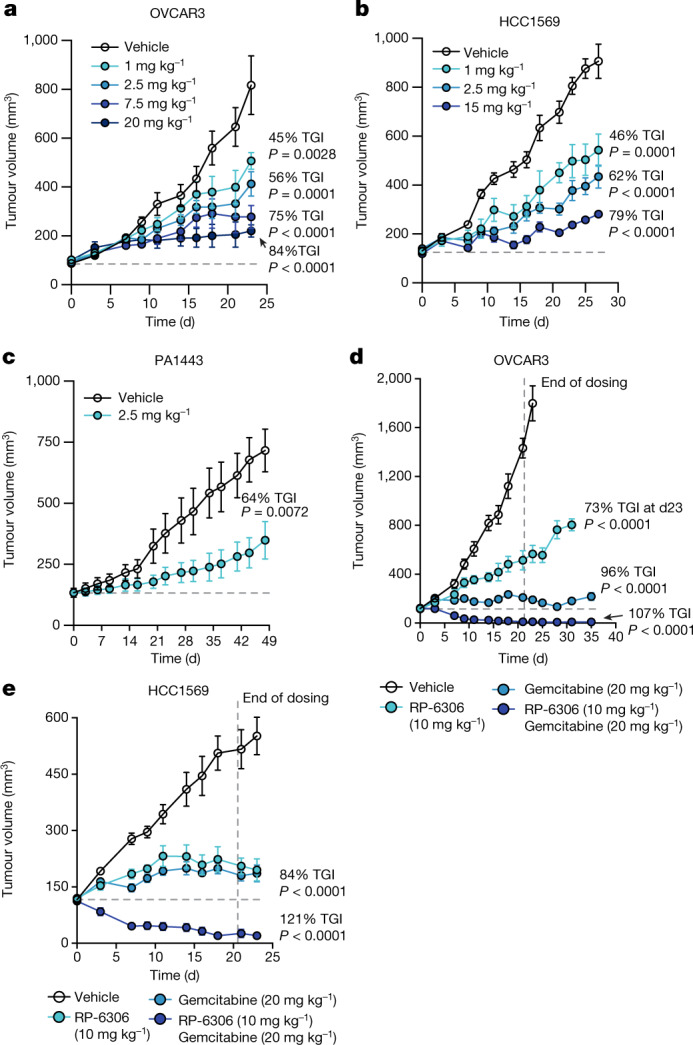
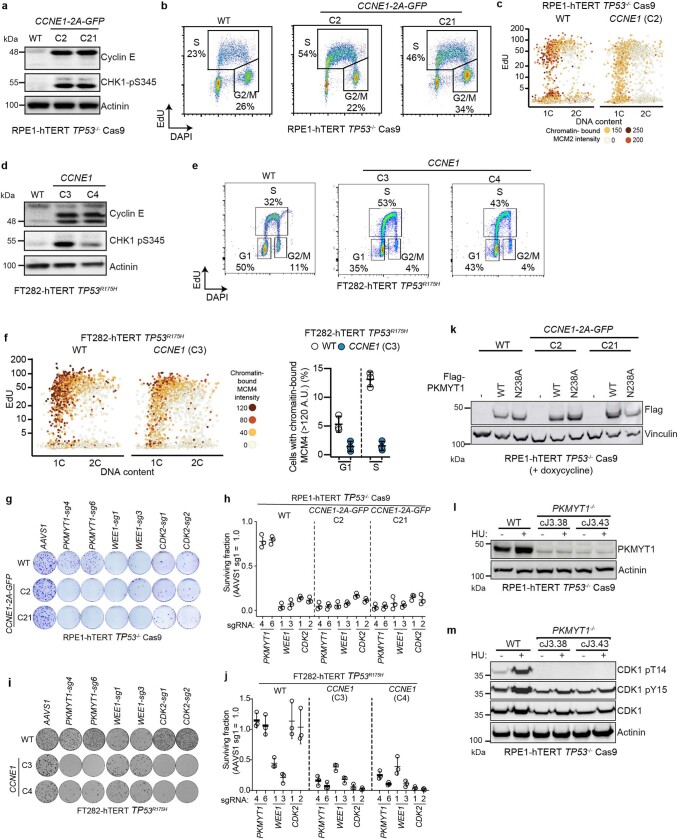
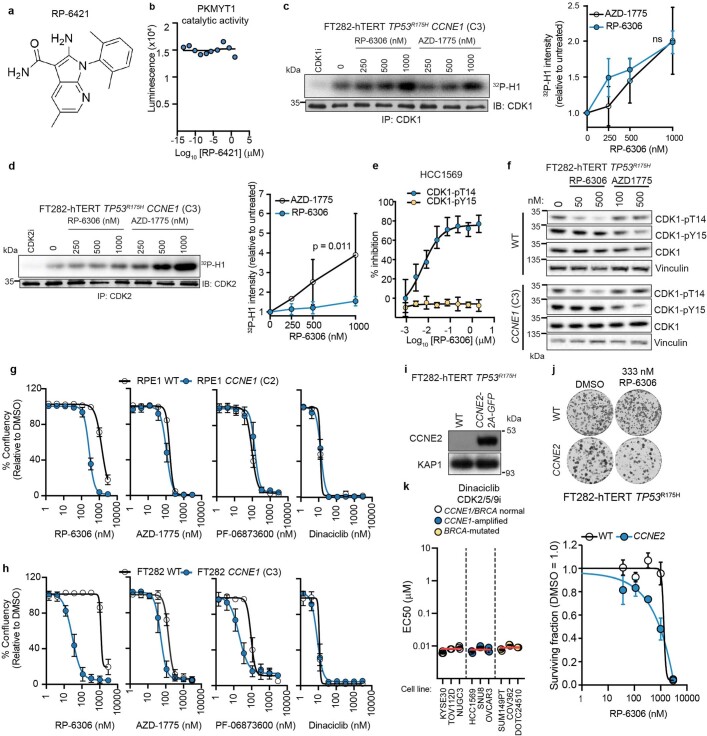
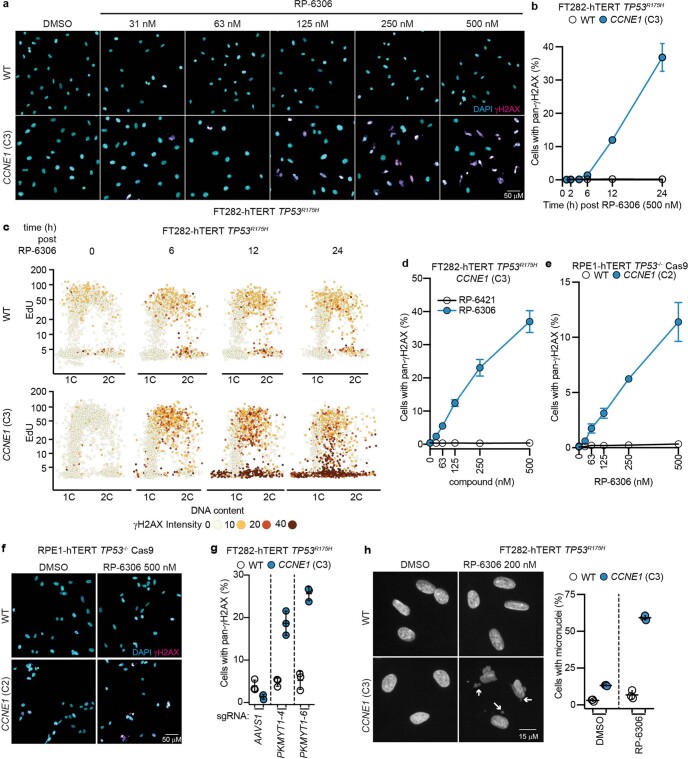
References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
