

Somatic genomic changes in single Alzheimer's disease neurons
- PMID: 35444284
- PMCID: PMC9357465
- DOI: 10.1038/s41586-022-04640-1
Somatic genomic changes in single Alzheimer's disease neurons
Abstract
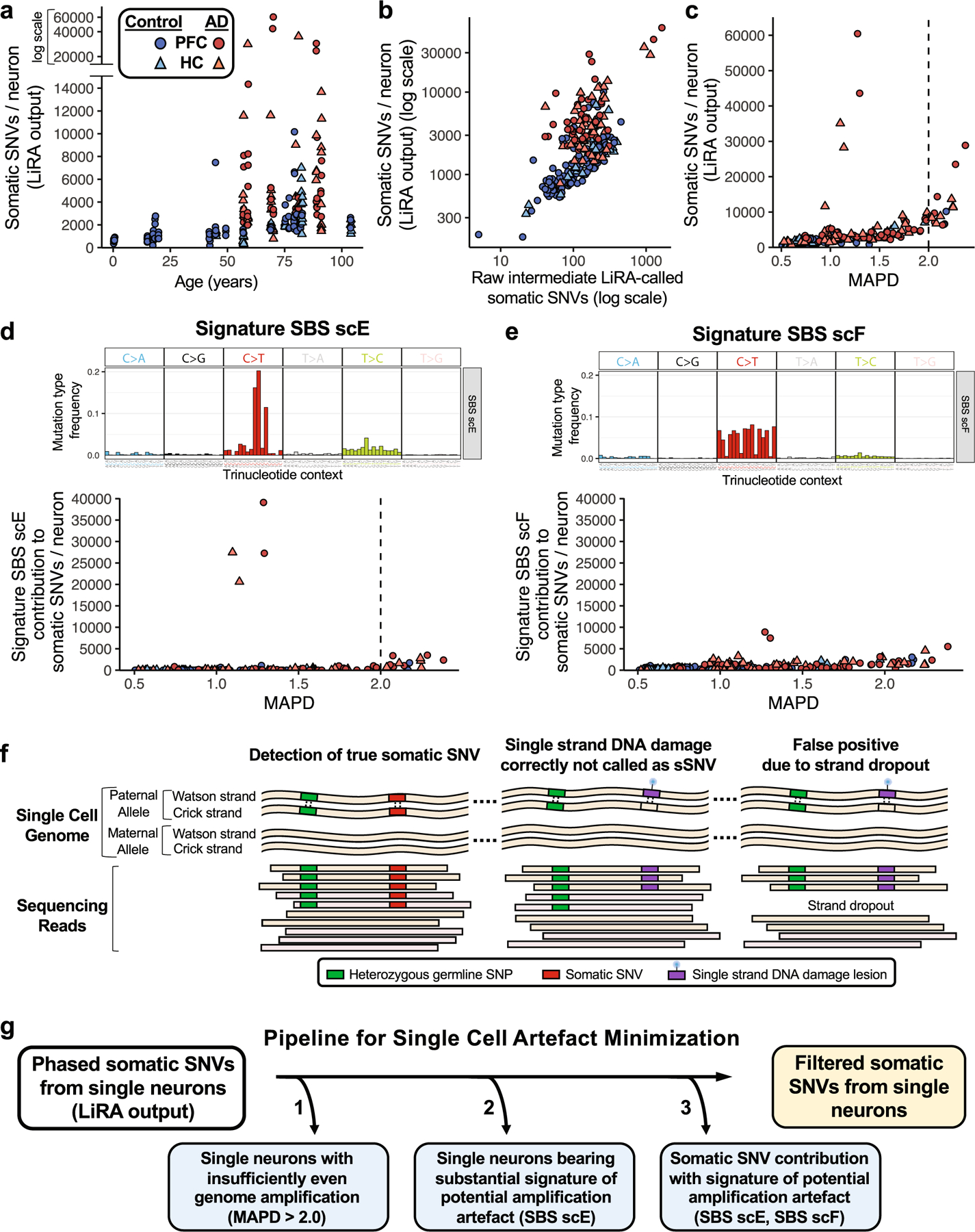
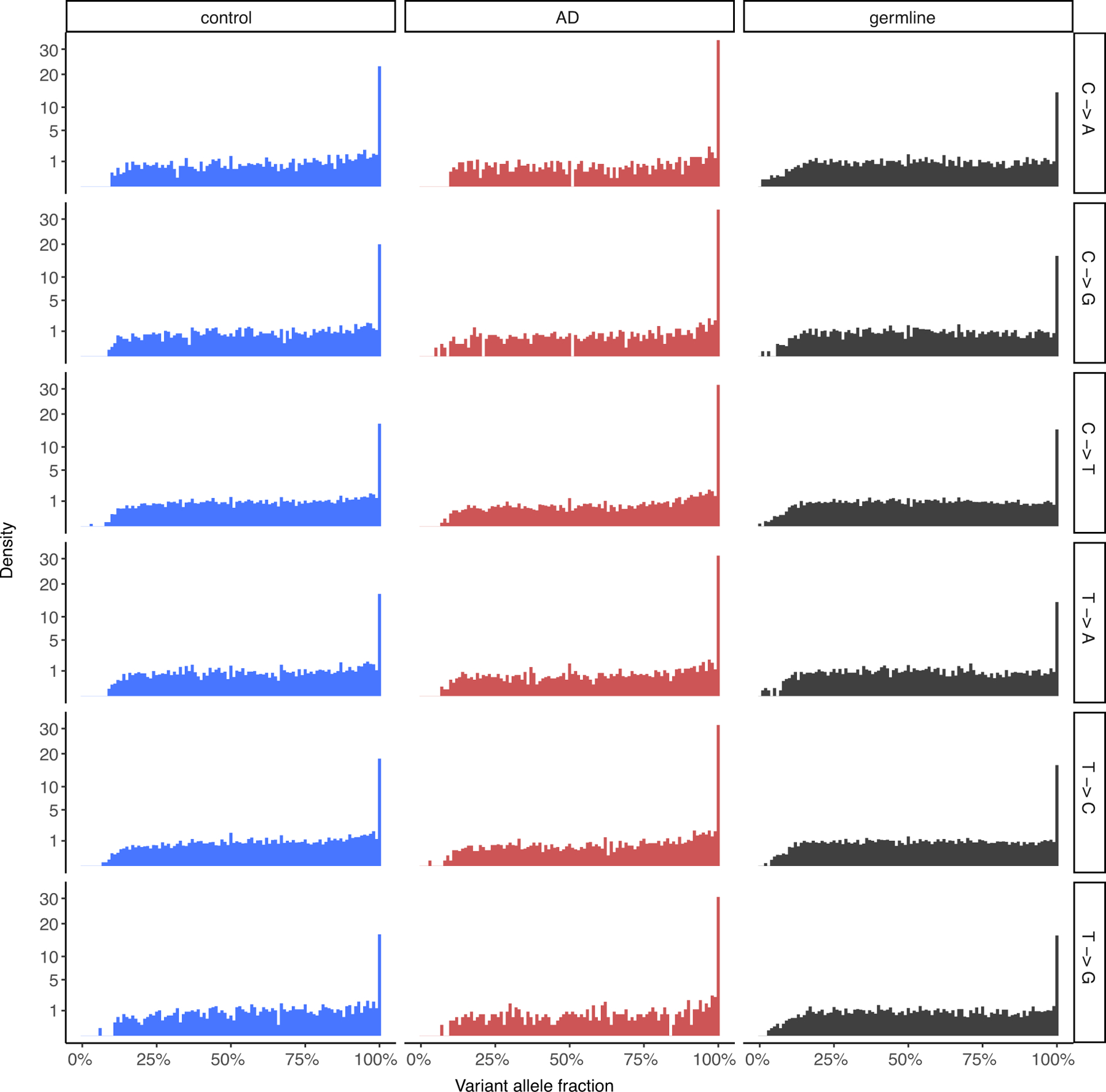
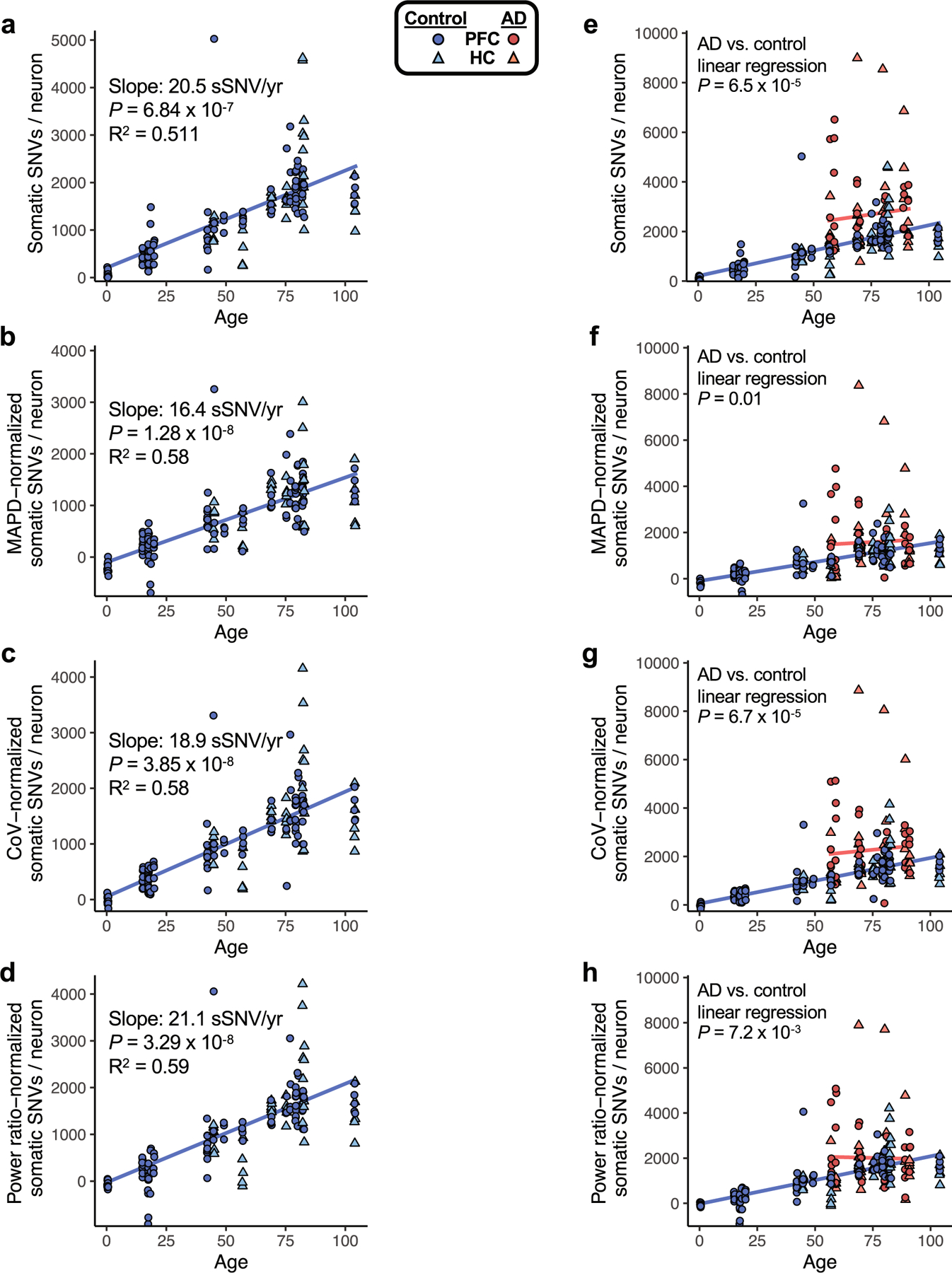
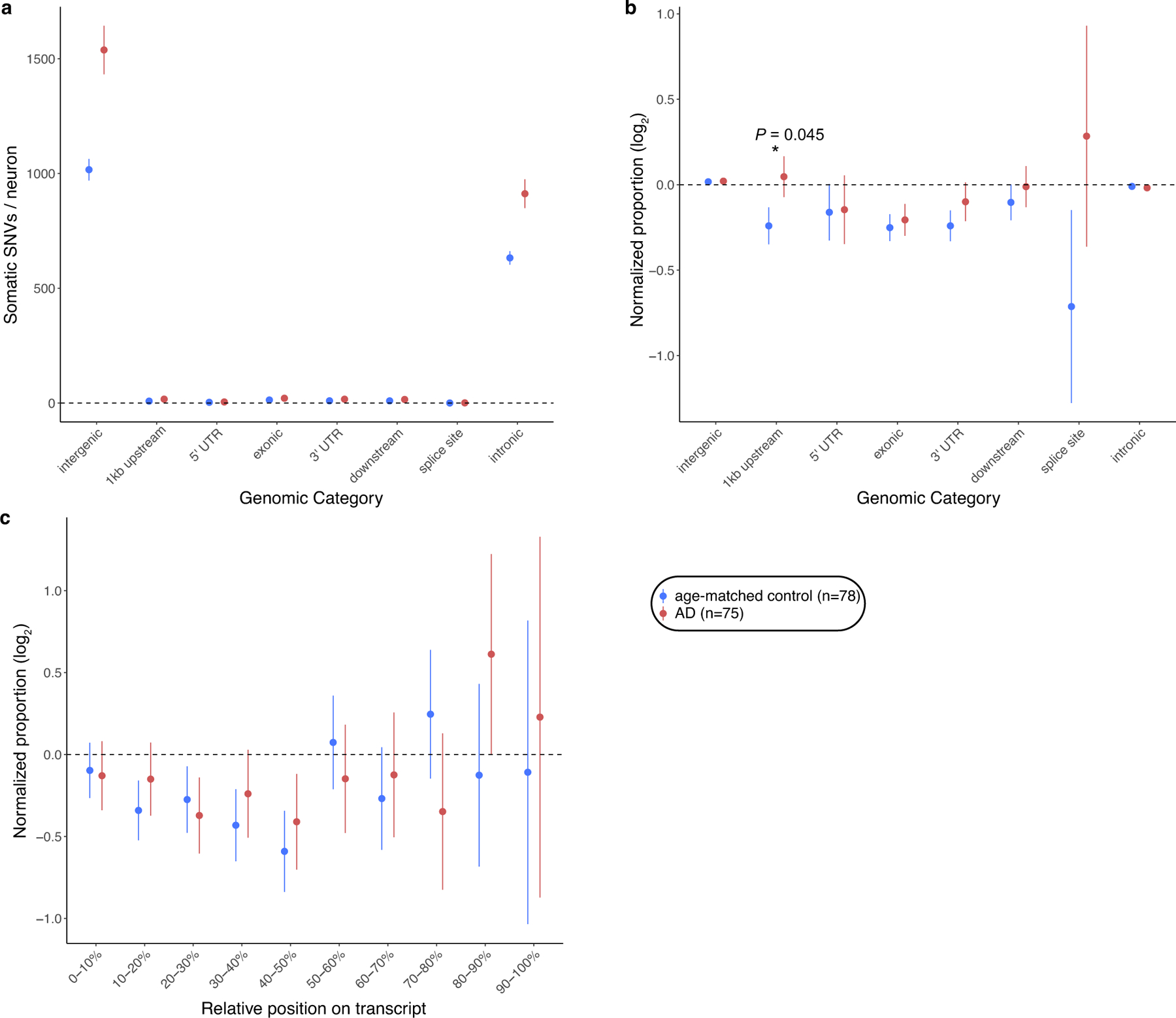
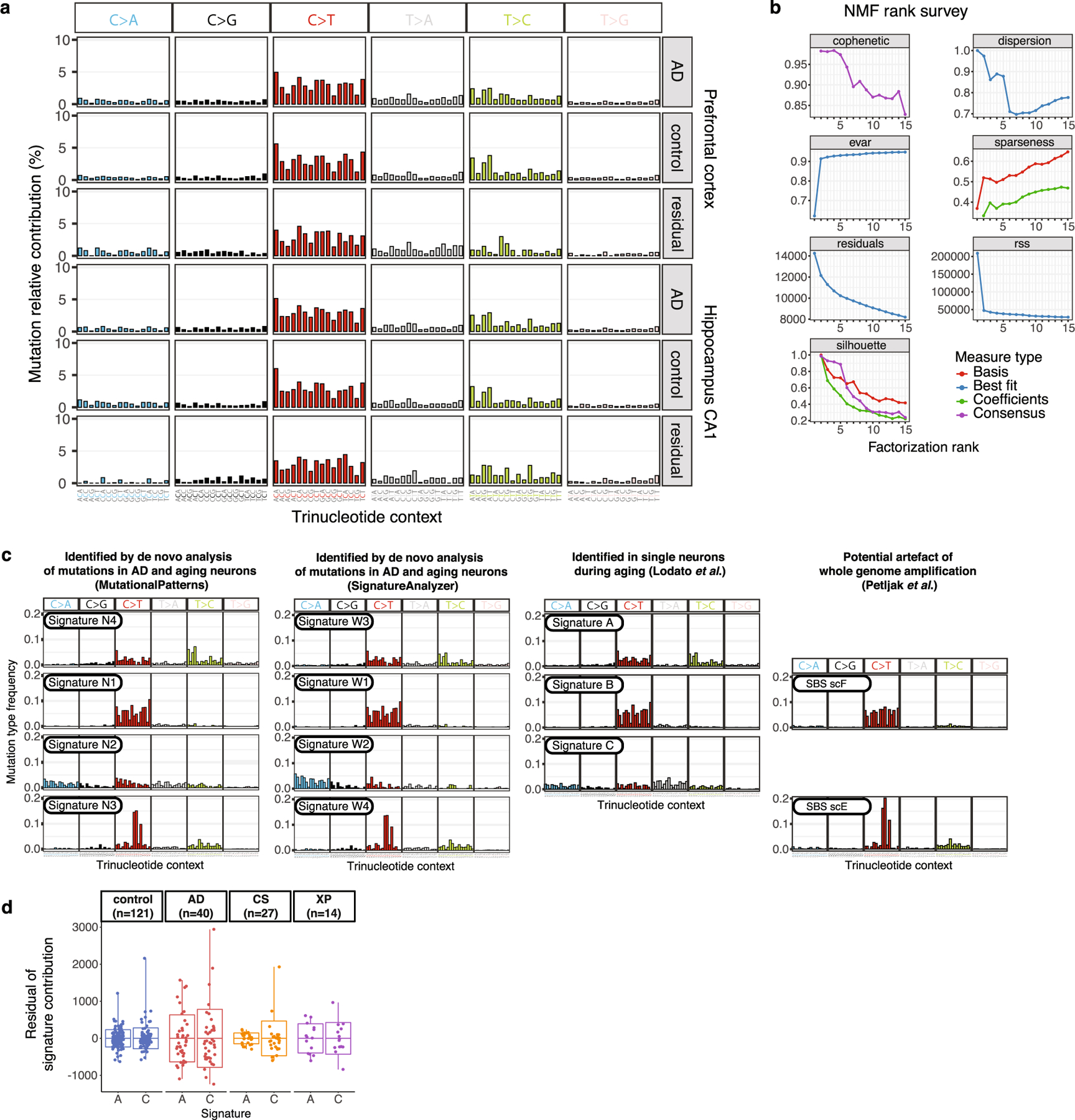
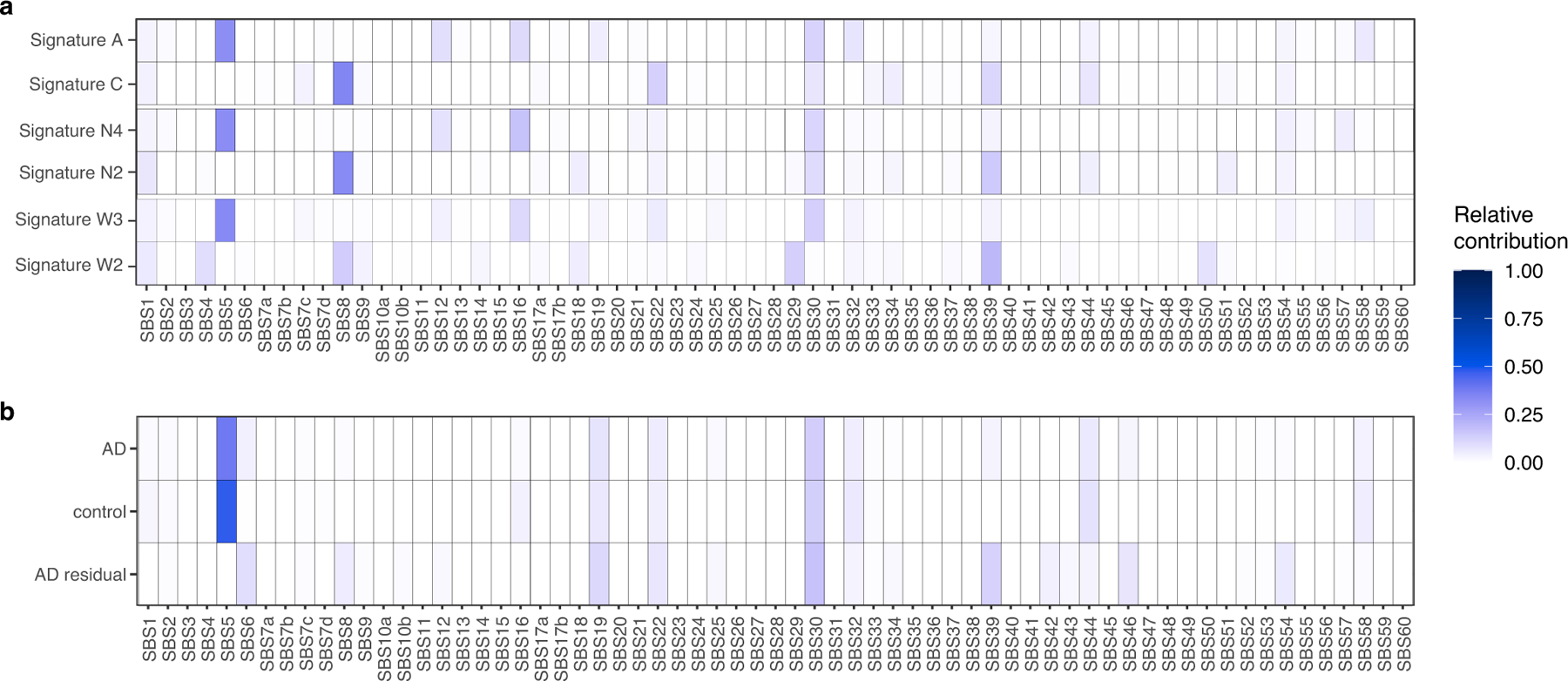
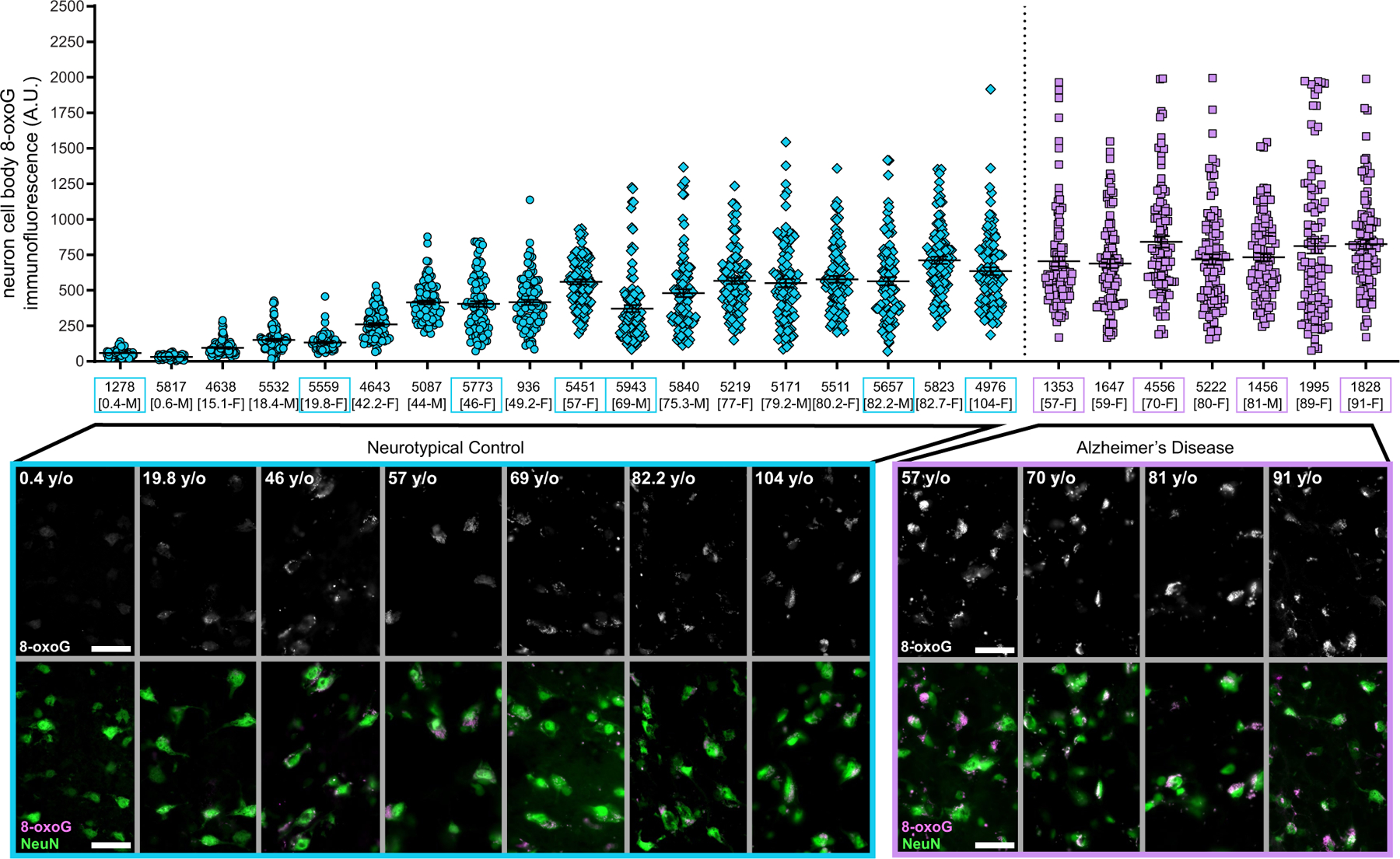
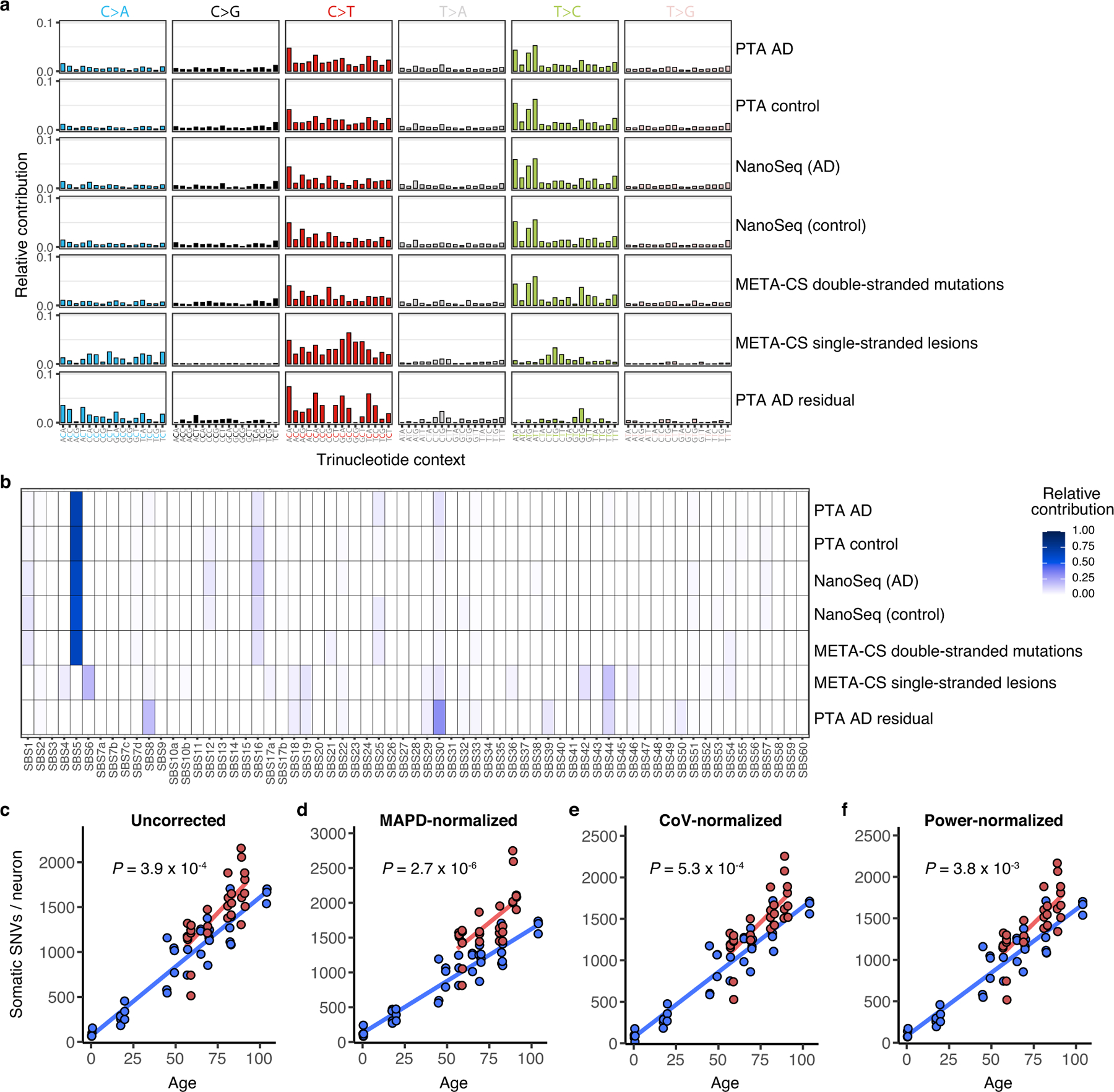
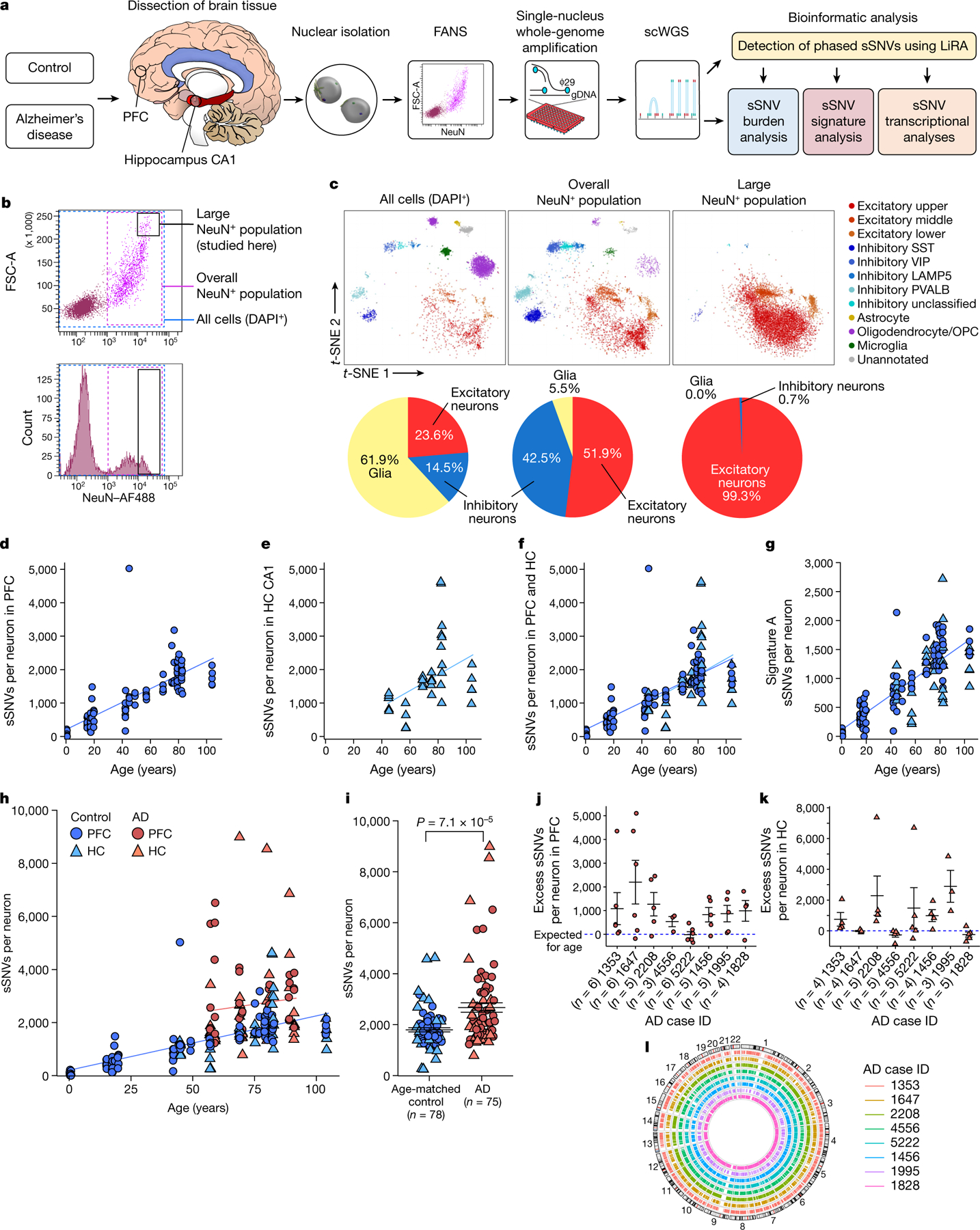
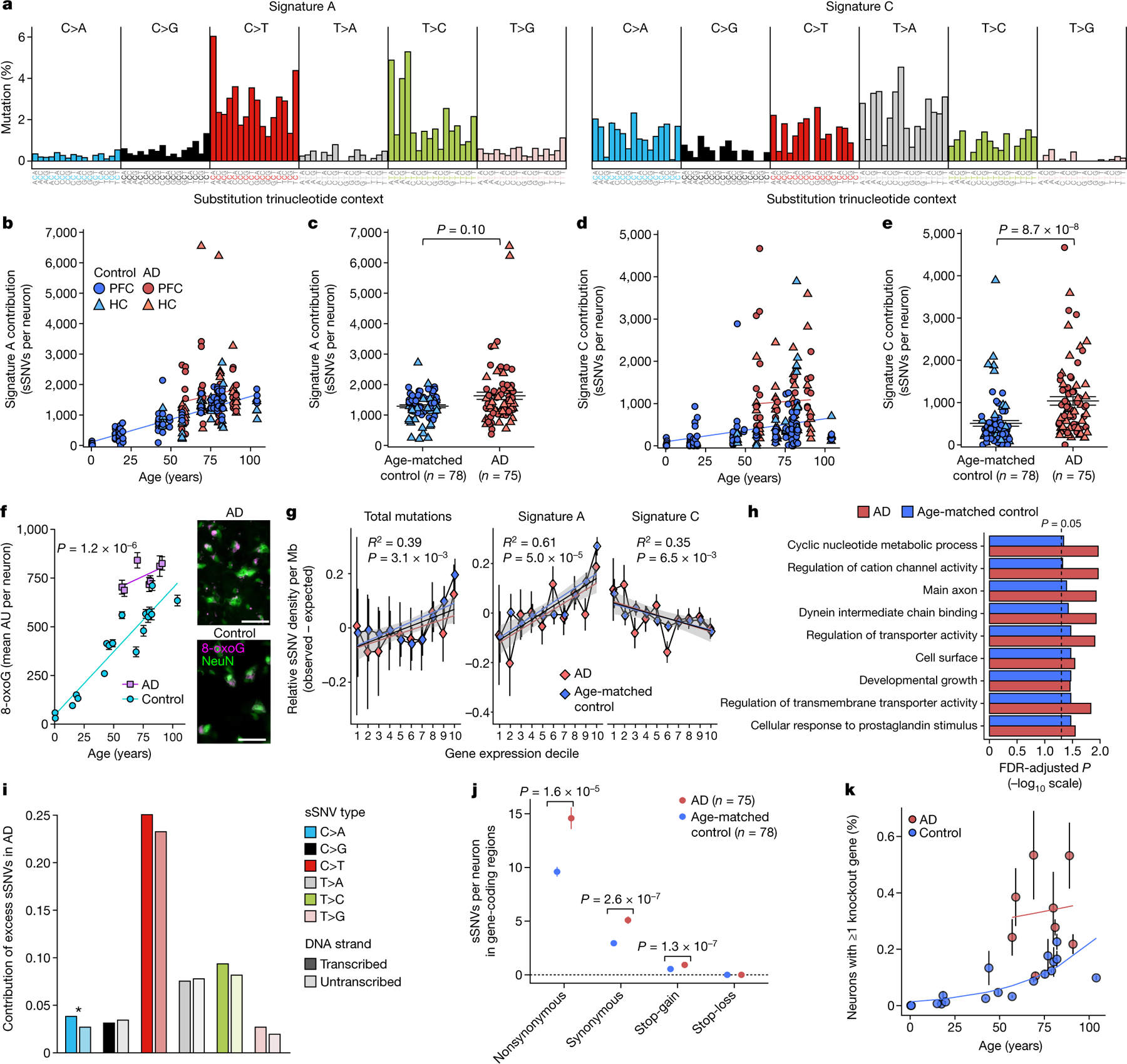
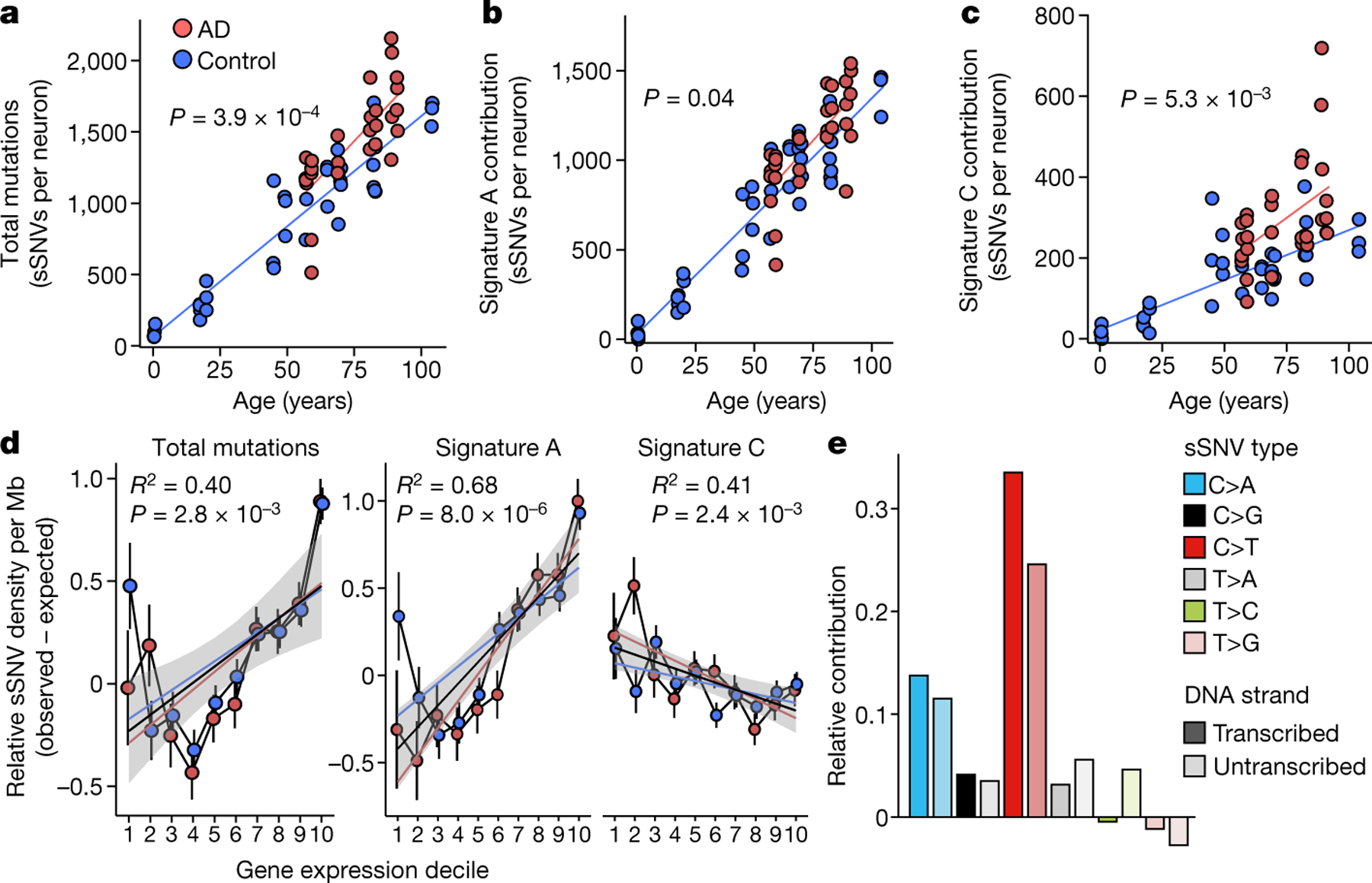
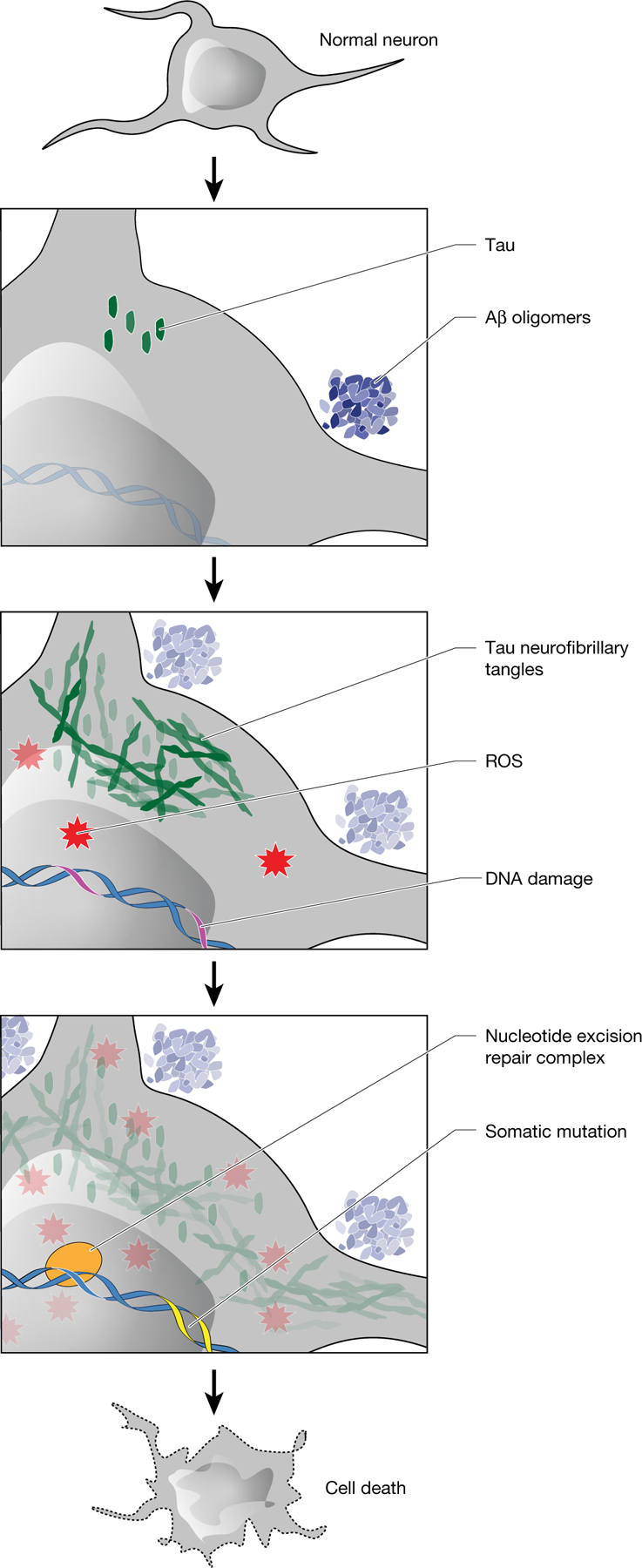
Dementia in Alzheimer's disease progresses alongside neurodegeneration1-4, but the specific events that cause neuronal dysfunction and death remain poorly understood. During normal ageing, neurons progressively accumulate somatic mutations5 at rates similar to those of dividing cells6,7 which suggests that genetic factors, environmental exposures or disease states might influence this accumulation5. Here we analysed single-cell whole-genome sequencing data from 319 neurons from the prefrontal cortex and hippocampus of individuals with Alzheimer's disease and neurotypical control individuals. We found that somatic DNA alterations increase in individuals with Alzheimer's disease, with distinct molecular patterns. Normal neurons accumulate mutations primarily in an age-related pattern (signature A), which closely resembles 'clock-like' mutational signatures that have been previously described in healthy and cancerous cells6-10. In neurons affected by Alzheimer's disease, additional DNA alterations are driven by distinct processes (signature C) that highlight C>A and other specific nucleotide changes. These changes potentially implicate nucleotide oxidation4,11, which we show is increased in Alzheimer's-disease-affected neurons in situ. Expressed genes exhibit signature-specific damage, and mutations show a transcriptional strand bias, which suggests that transcription-coupled nucleotide excision repair has a role in the generation of mutations. The alterations in Alzheimer's disease affect coding exons and are predicted to create dysfunctional genetic knockout cells and proteostatic stress. Our results suggest that known pathogenic mechanisms in Alzheimer's disease may lead to genomic damage to neurons that can progressively impair function. The aberrant accumulation of DNA alterations in neurodegeneration provides insight into the cascade of molecular and cellular events that occurs in the development of Alzheimer's disease.
© 2022. The Author(s), under exclusive licence to Springer Nature Limited.
Conflict of interest statement
Figures
References
MeSH terms
Substances
Grants and funding
- HHMI/Howard Hughes Medical Institute/United States
- K08 AG065502/AG/NIA NIH HHS/United States
- K01 AG051791/AG/NIA NIH HHS/United States
- R01 NS032457/NS/NINDS NIH HHS/United States
- R00 AG054748/AG/NIA NIH HHS/United States
- P50 HD105351/HD/NICHD NIH HHS/United States
- T32 HL007627/HL/NHLBI NIH HHS/United States
- P30 AG062421/AG/NIA NIH HHS/United States
- DP2 AG072437/AG/NIA NIH HHS/United States
- T32 HG002295/HG/NHGRI NIH HHS/United States
- T32 GM007753/GM/NIGMS NIH HHS/United States
- R01 AG070921/AG/NIA NIH HHS/United States
- T15 LM007098/NH/NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
