

Inhibition of the NOTCH1 Pathway in the Stressed Heart Limits Fibrosis and Promotes Recruitment of Non-Myocyte Cells into the Cardiomyocyte Fate
- PMID: 35448087
- PMCID: PMC9024539
- DOI: 10.3390/jcdd9040111
Inhibition of the NOTCH1 Pathway in the Stressed Heart Limits Fibrosis and Promotes Recruitment of Non-Myocyte Cells into the Cardiomyocyte Fate
Abstract
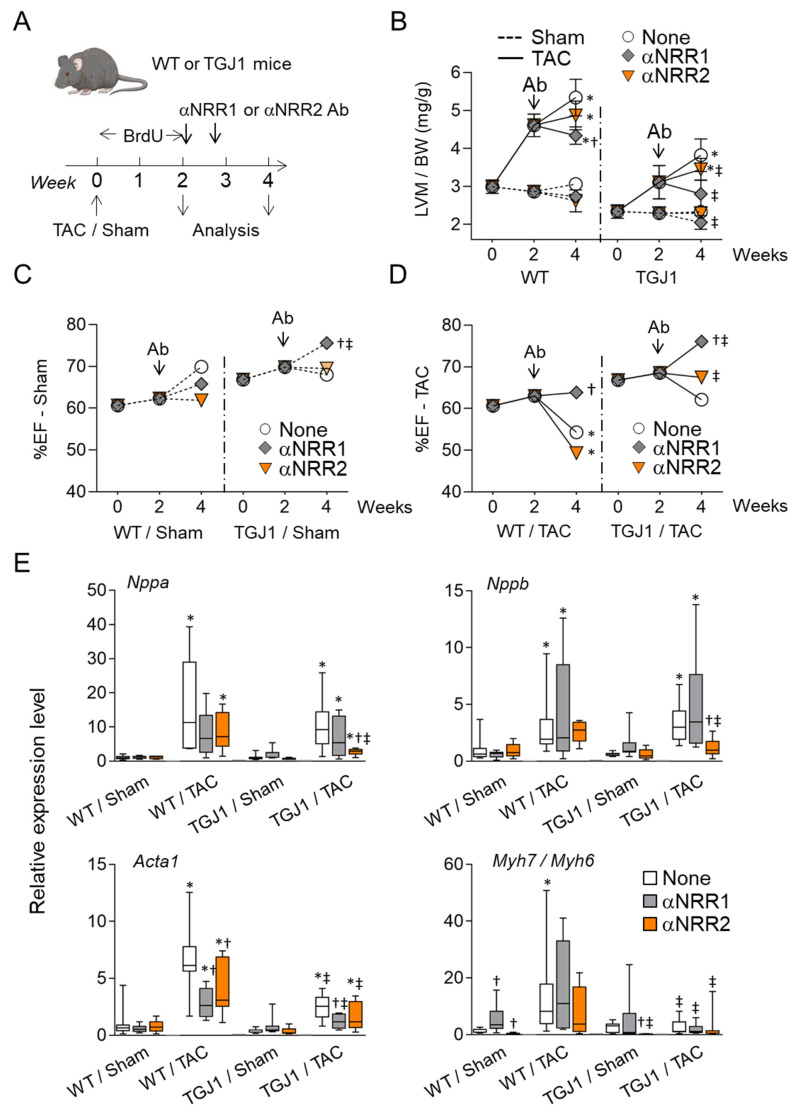
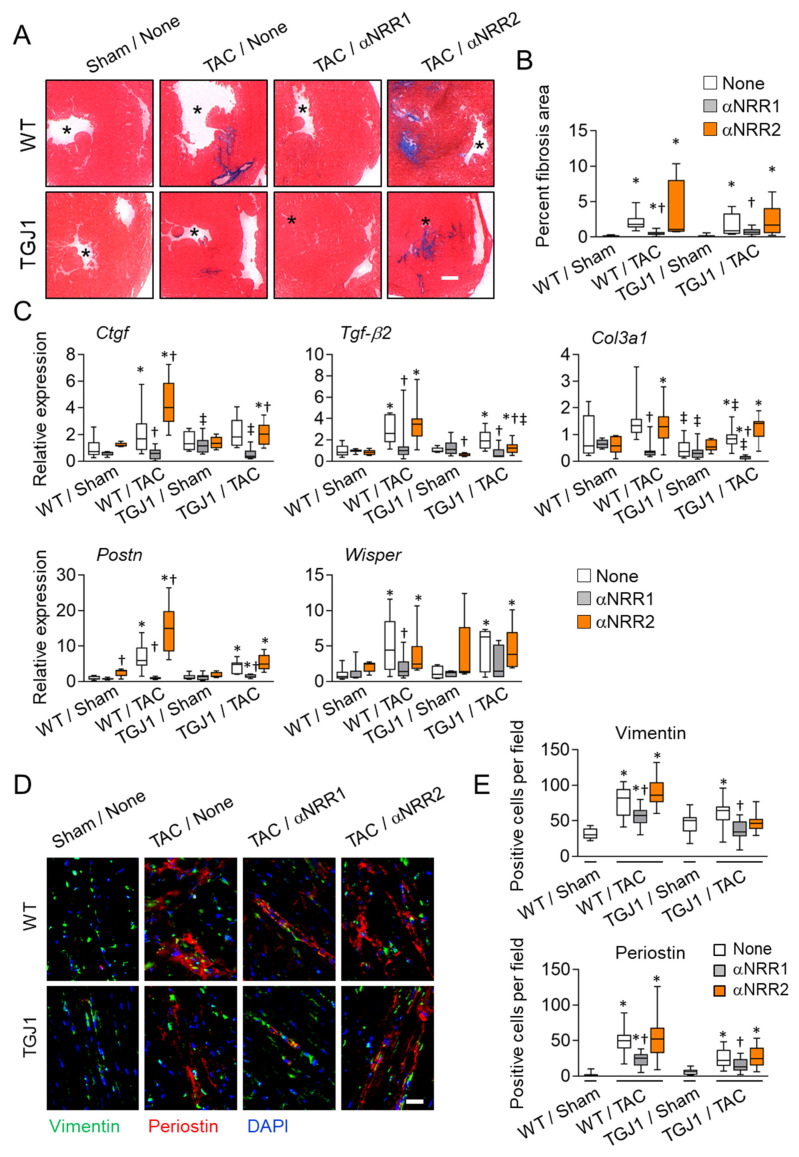
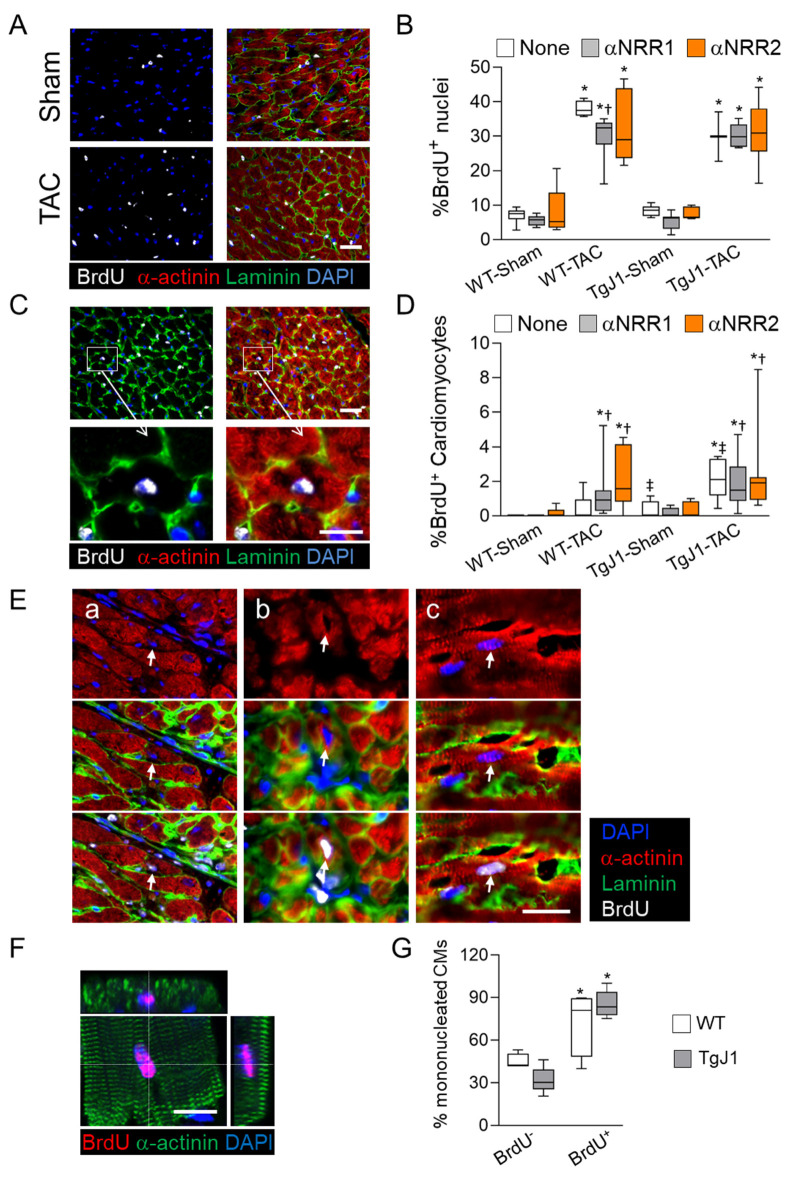
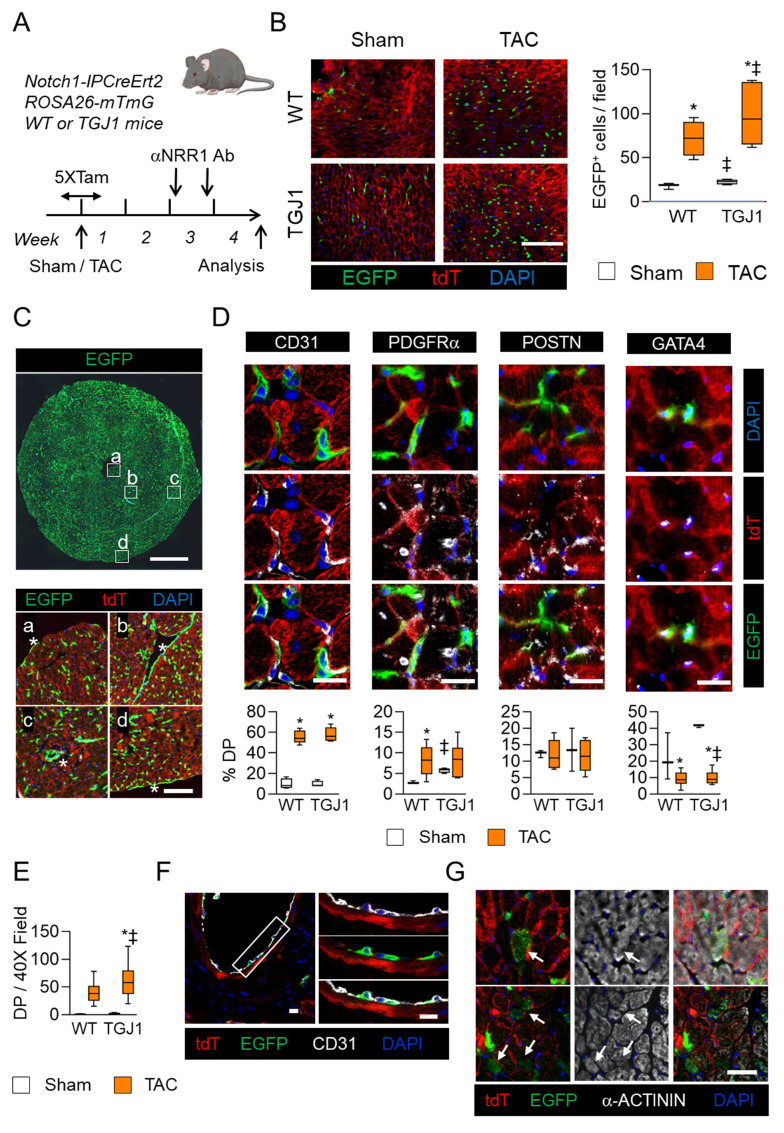
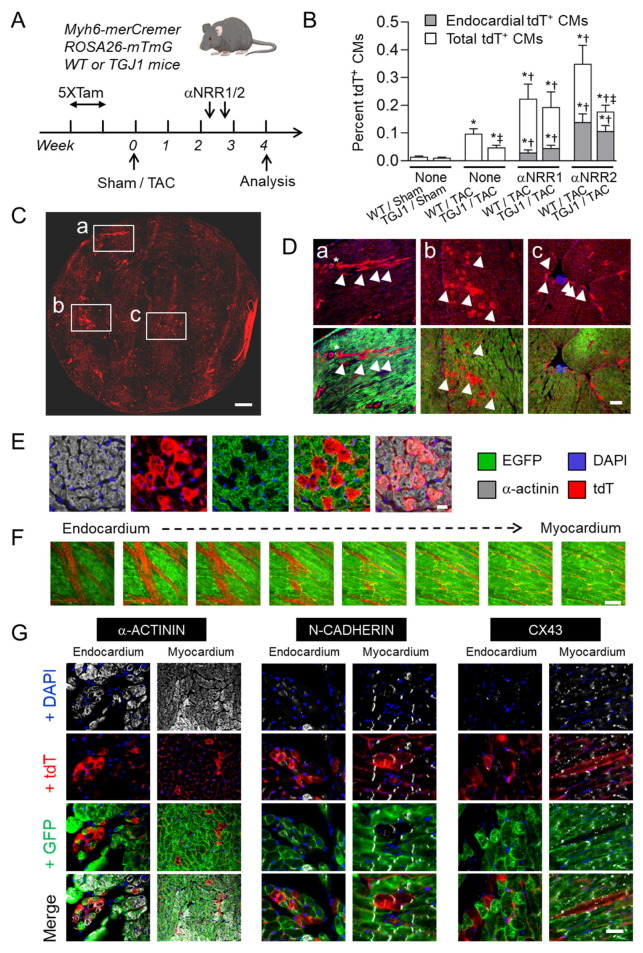
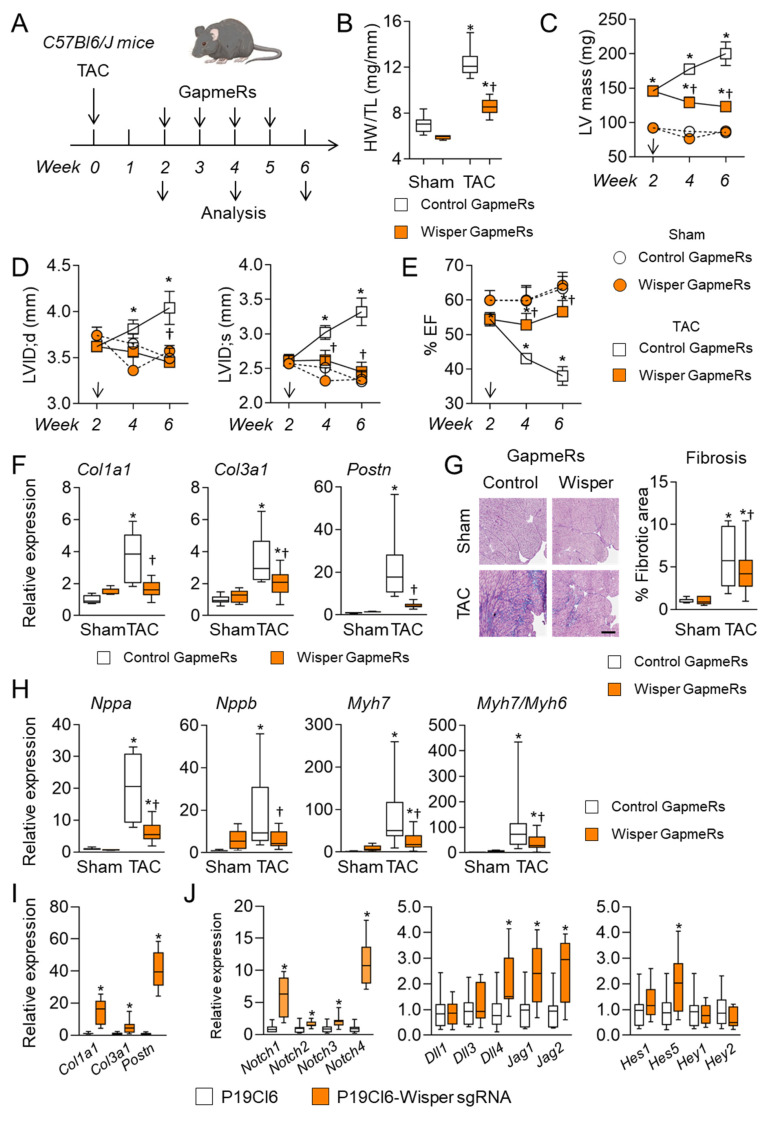
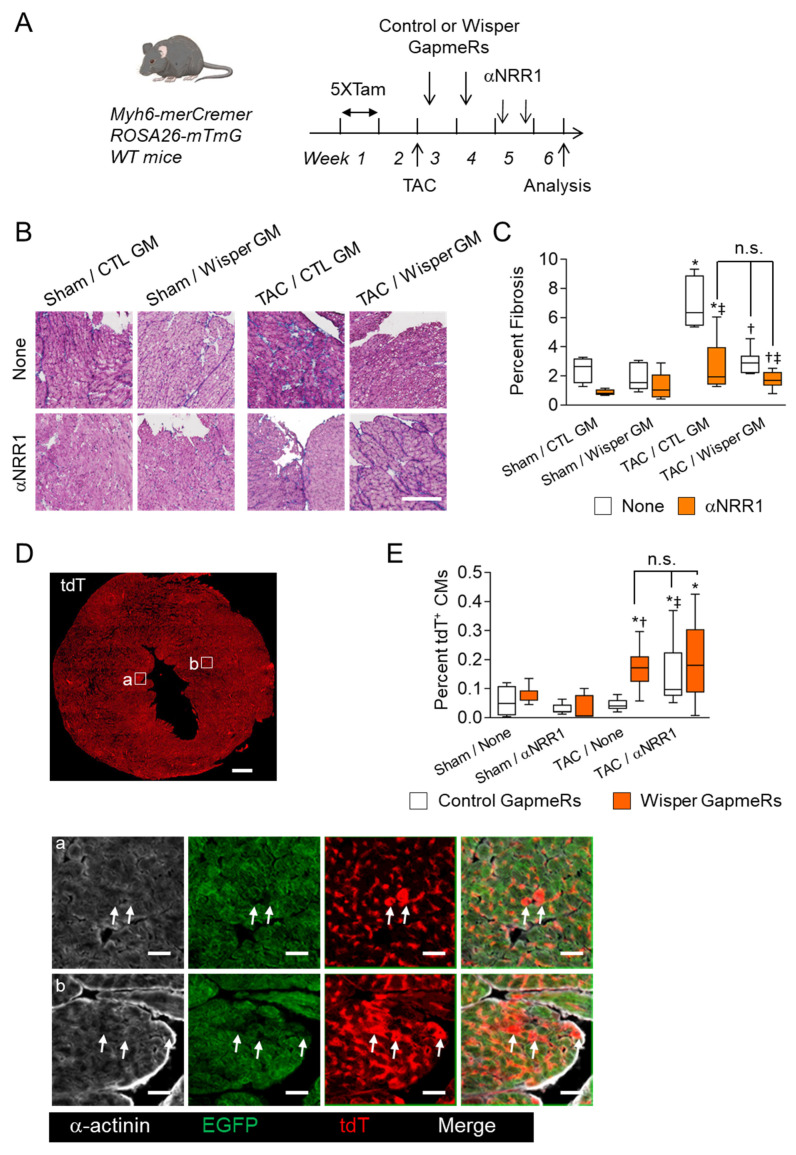
Cardiac pathologies lead to an acute or gradual loss of cardiomyocytes. Because of the limited regenerative capacity of the mammalian heart, cardiomyocytes are only replaced by fibrotic tissue. Excessive fibrosis contributes to the deterioration of cardiac function and the transition to heart failure, which is the leading cause of morbidity and mortality worldwide. Currently, no treatments can promote replenishment of the injured heart with newly formed cardiomyocytes. In this context, regenerative strategies explore the possibility to promote recovery through induction of cardiomyocyte production from pre-existing cardiomyocytes. On the other hand, cardiac non-myocyte cells can be directly reprogrammed into induced cardiac precursor cells and cardiomyocytes, suggesting that these cells could be exploited to produce cardiomyocytes in vivo. Here, we provide evidence that the sequential activation and inhibition of the NOTCH1 signaling pathway in the stressed heart decreases fibrosis and improves cardiac function in the stressed heart. This is accompanied by the emergence of new cardiomyocytes from non-myocyte origin. Overall, our data show how a developmental pathway such as the NOTCH pathway can be manipulated to provide therapeutic benefit in the damaged heart.
Keywords: NOTCH; cardiomyocyte production; fibrosis; heart; remodeling.
Conflict of interest statement
TP is a co-founder of Haya Therapeutics, Lausanne, Switzerland. The other authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
Figures
Similar articles
-
The Notch pathway controls fibrotic and regenerative repair in the adult heart.Eur Heart J. 2014 Aug 21;35(32):2174-85. doi: 10.1093/eurheartj/ehs269. Epub 2012 Nov 19. Eur Heart J. 2014. PMID: 23166366 Free PMC article.
-
Control of the adaptive response of the heart to stress via the Notch1 receptor pathway.J Exp Med. 2008 Dec 22;205(13):3173-85. doi: 10.1084/jem.20081427. Epub 2008 Dec 8. J Exp Med. 2008. PMID: 19064701 Free PMC article.
-
Jagged1 intracellular domain-mediated inhibition of Notch1 signalling regulates cardiac homeostasis in the postnatal heart.Cardiovasc Res. 2015 Oct 1;108(1):74-86. doi: 10.1093/cvr/cvv209. Epub 2015 Aug 6. Cardiovasc Res. 2015. PMID: 26249804 Free PMC article.
-
Cardiac Fibroblasts and Myocardial Regeneration.Front Bioeng Biotechnol. 2021 Mar 25;9:599928. doi: 10.3389/fbioe.2021.599928. eCollection 2021. Front Bioeng Biotechnol. 2021. PMID: 33842440 Free PMC article. Review.
-
Leading progress in heart regeneration and repair.Curr Opin Cell Biol. 2019 Dec;61:79-85. doi: 10.1016/j.ceb.2019.07.005. Epub 2019 Aug 10. Curr Opin Cell Biol. 2019. PMID: 31408771 Free PMC article. Review.
Cited by
-
Effects of maternal hypothyroidism on postnatal cardiomyocyte proliferation and cardiac disease responses of the progeny.Am J Physiol Heart Circ Physiol. 2023 Oct 1;325(4):H702-H719. doi: 10.1152/ajpheart.00320.2023. Epub 2023 Aug 4. Am J Physiol Heart Circ Physiol. 2023. PMID: 37539452 Free PMC article.
-
Research Advances in Myocardial Infarction Repair and Cardiac Regenerative Medicine via the Notch Signaling Pathway.Rev Cardiovasc Med. 2025 Mar 19;26(3):26587. doi: 10.31083/RCM26587. eCollection 2025 Mar. Rev Cardiovasc Med. 2025. PMID: 40160574 Free PMC article. Review.
-
Direct Reprogramming of Resident Non-Myocyte Cells and Its Potential for In Vivo Cardiac Regeneration.Cells. 2023 Apr 15;12(8):1166. doi: 10.3390/cells12081166. Cells. 2023. PMID: 37190075 Free PMC article. Review.
-
Inhibiting AGTR1 reduces AML burden and protects the heart from cardiotoxicity in mouse models.Sci Transl Med. 2024 Jun 19;16(752):eadl5931. doi: 10.1126/scitranslmed.adl5931. Epub 2024 Jun 19. Sci Transl Med. 2024. PMID: 38896605 Free PMC article.
References
-
- Ali S.R., Ranjbarvaziri S., Talkhabi M., Zhao P., Subat A., Hojjat A., Kamran P., Muller A.M., Volz K.S., Tang Z., et al. Developmental heterogeneity of cardiac fibroblasts does not predict pathological proliferation and activation. Circ. Res. 2014;115:625–635. doi: 10.1161/CIRCRESAHA.115.303794. - DOI - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
