

DestVI identifies continuums of cell types in spatial transcriptomics data
- PMID: 35449415
- PMCID: PMC9756396
- DOI: 10.1038/s41587-022-01272-8
DestVI identifies continuums of cell types in spatial transcriptomics data
Abstract
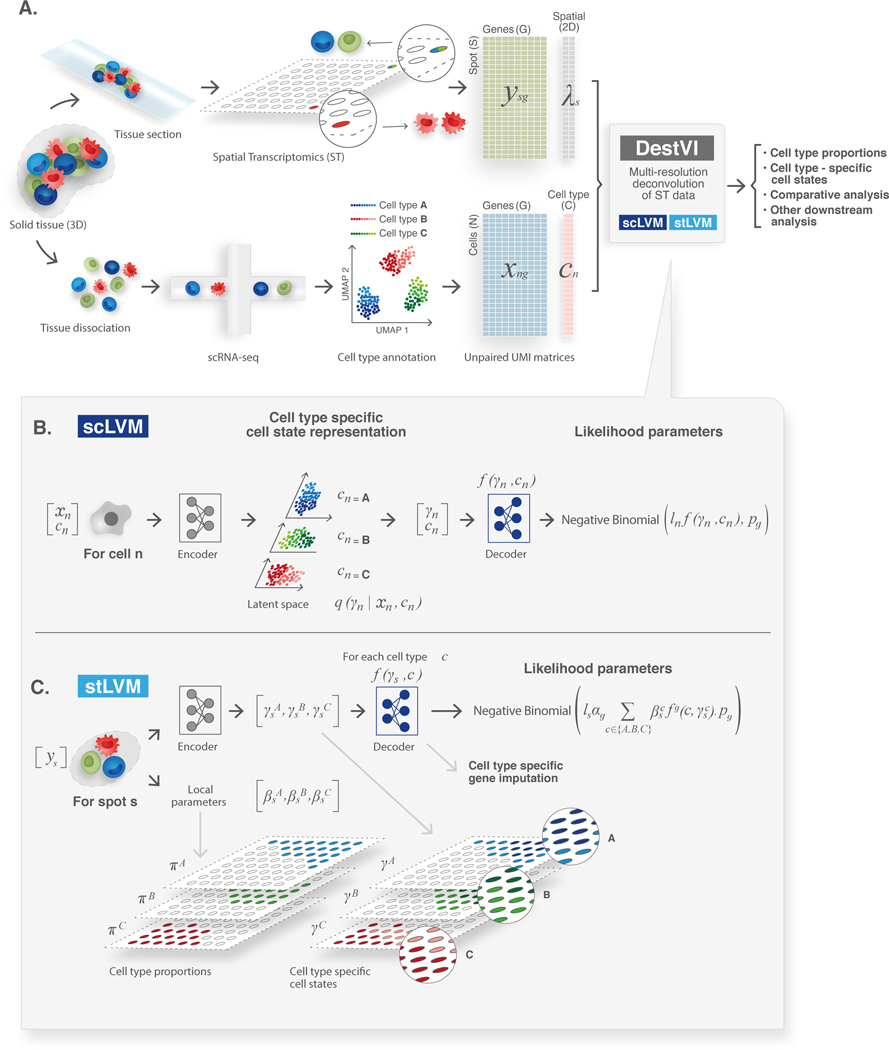
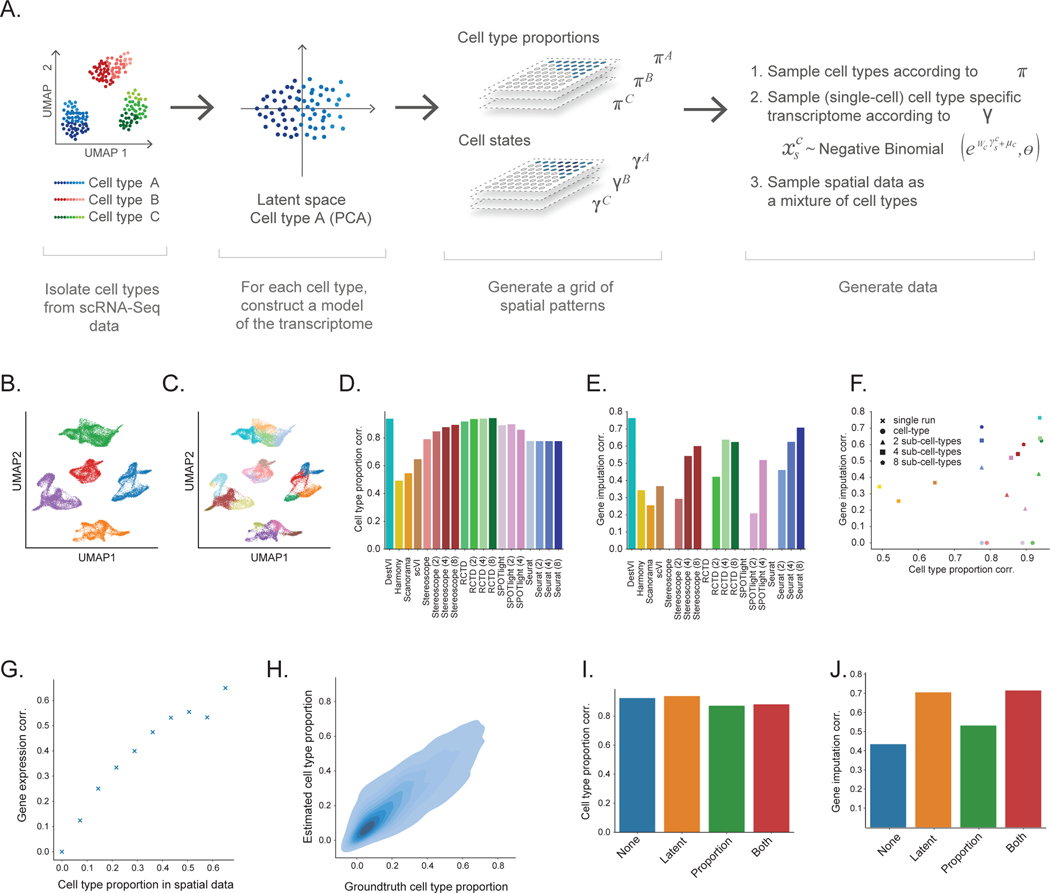
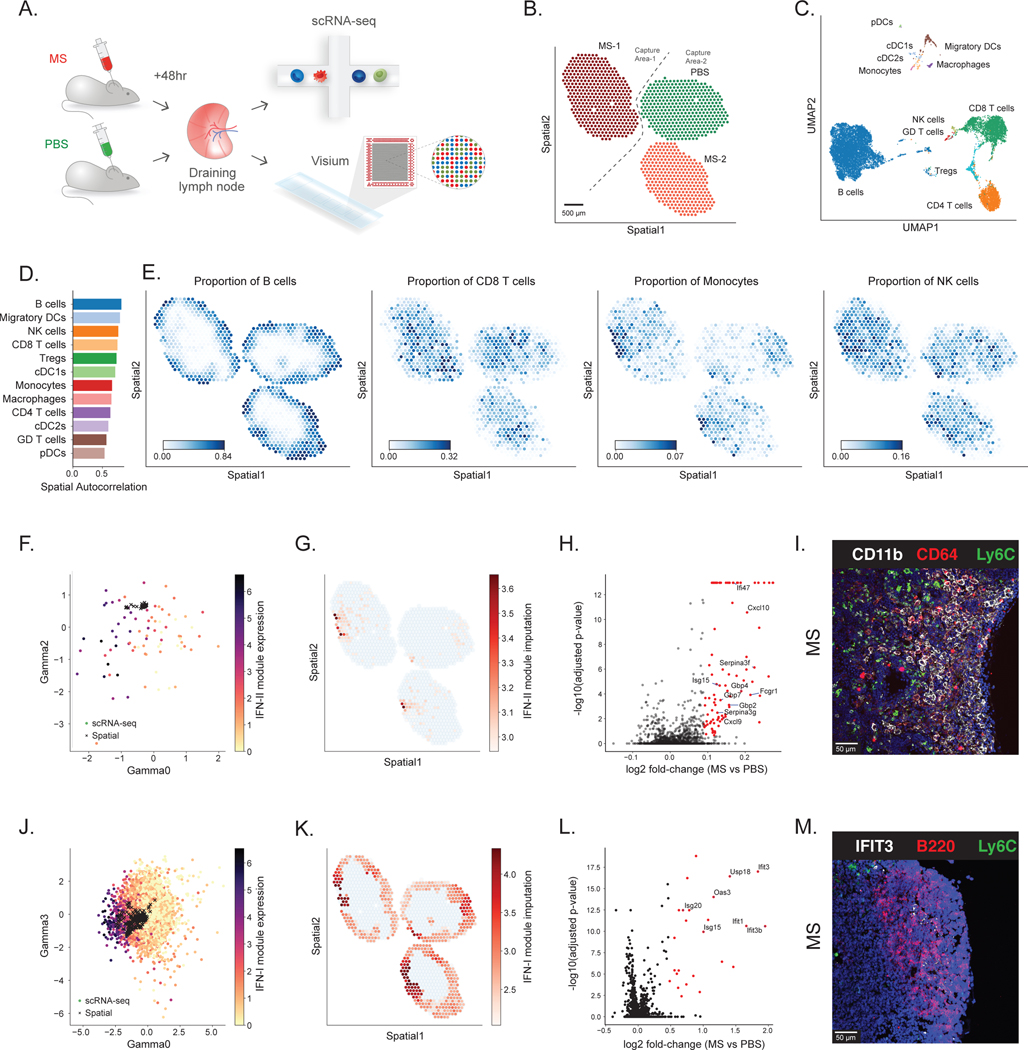
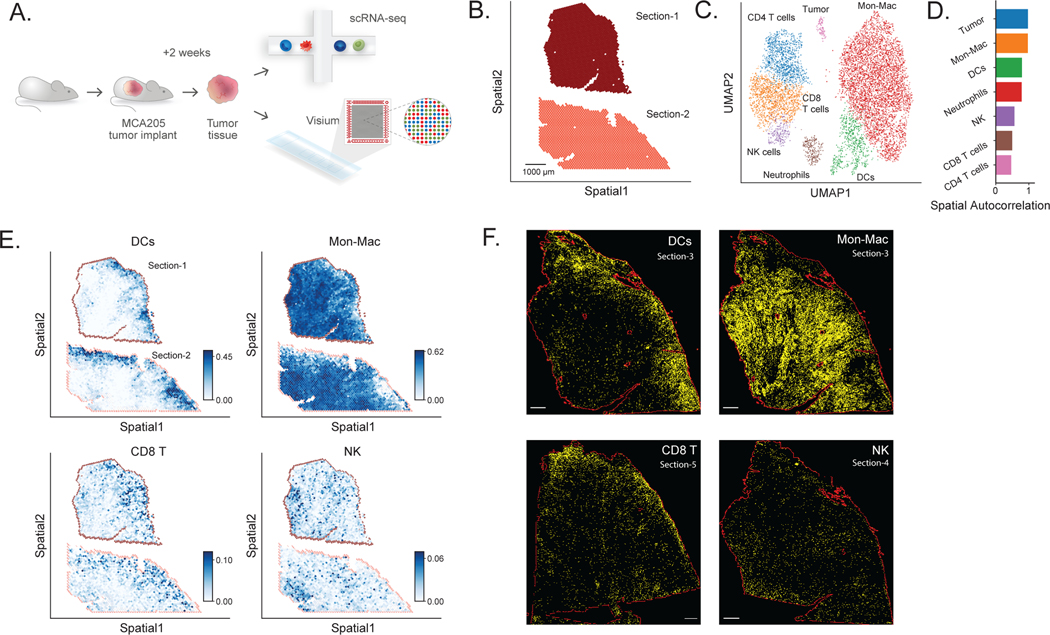
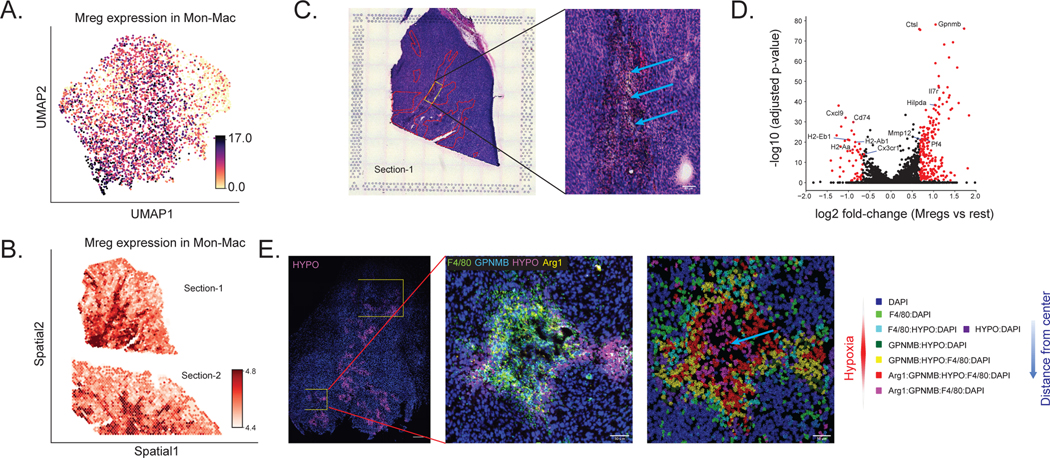
Most spatial transcriptomics technologies are limited by their resolution, with spot sizes larger than that of a single cell. Although joint analysis with single-cell RNA sequencing can alleviate this problem, current methods are limited to assessing discrete cell types, revealing the proportion of cell types inside each spot. To identify continuous variation of the transcriptome within cells of the same type, we developed Deconvolution of Spatial Transcriptomics profiles using Variational Inference (DestVI). Using simulations, we demonstrate that DestVI outperforms existing methods for estimating gene expression for every cell type inside every spot. Applied to a study of infected lymph nodes and of a mouse tumor model, DestVI provides high-resolution, accurate spatial characterization of the cellular organization of these tissues and identifies cell-type-specific changes in gene expression between different tissue regions or between conditions. DestVI is available as part of the open-source software package scvi-tools ( https://scvi-tools.org ).
© 2022. The Author(s), under exclusive licence to Springer Nature America, Inc.
Conflict of interest statement
Competing Interests Statement
N.Y. is an advisor and/or has equity in Cellarity, Celsius Therapeutics, and Rheos Medicine.
Figures
References
-
- Hunter MV, Moncada R, Weiss JM, Yanai I, White RM. Spatial transcriptomics reveals the architecture of the tumor/microenvironment interface. Cold Spring Harbor Laboratory. 2020. p. 2020.11.05.368753. doi:10.1101/2020.11.05.368753 - DOI
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
