

The clinical manifestations, molecular mechanisms and treatment of craniosynostosis
- PMID: 35451466
- PMCID: PMC9044212
- DOI: 10.1242/dmm.049390
The clinical manifestations, molecular mechanisms and treatment of craniosynostosis
Abstract
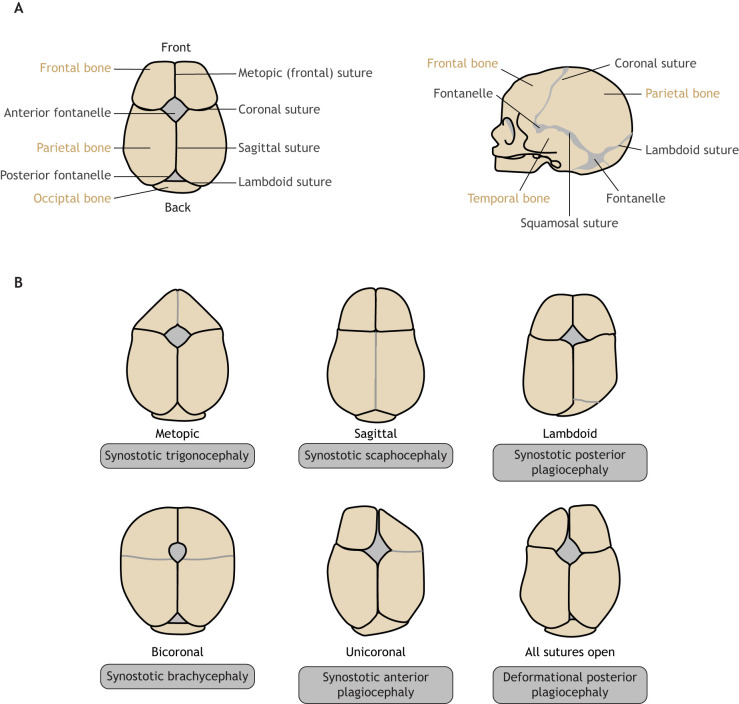
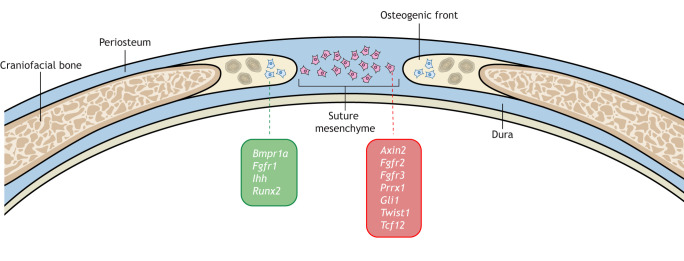
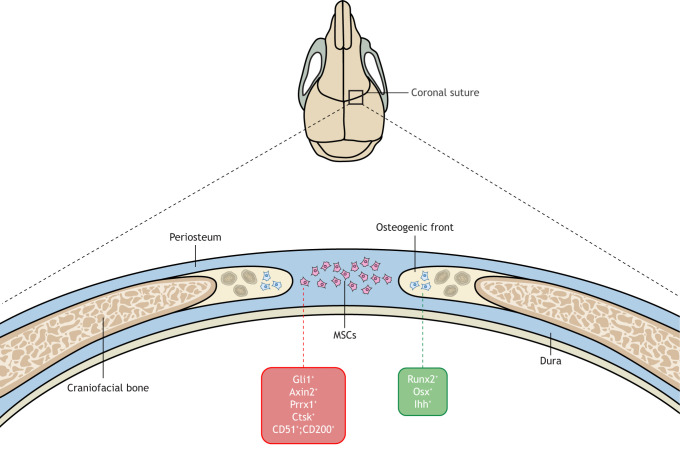
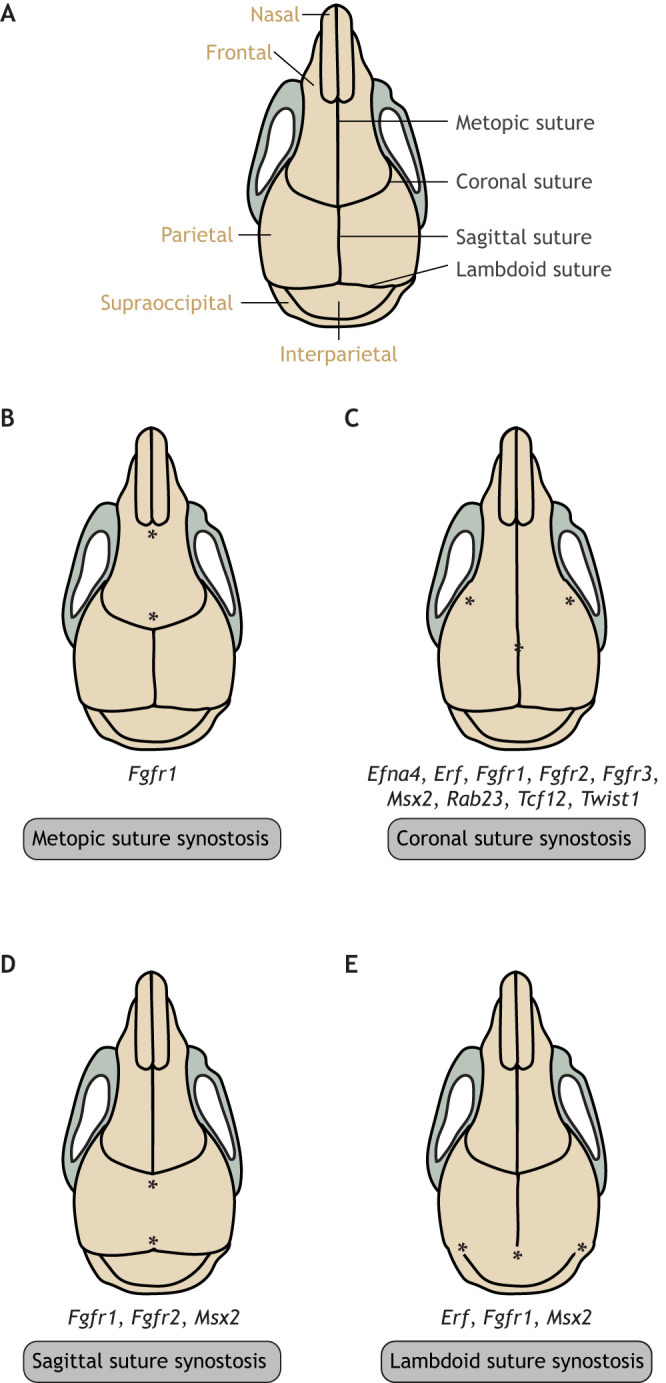
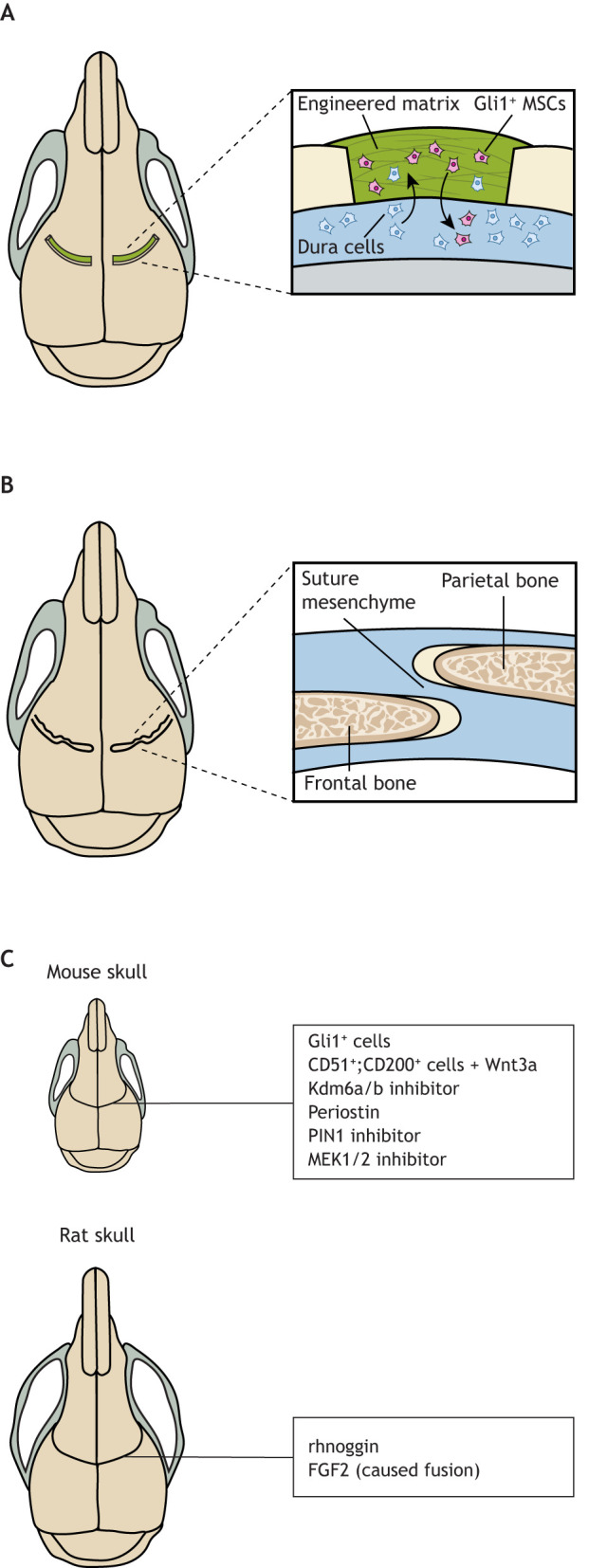
Craniosynostosis is a major congenital craniofacial disorder characterized by the premature fusion of cranial suture(s). Patients with severe craniosynostosis often have impairments in hearing, vision, intracranial pressure and/or neurocognitive functions. Craniosynostosis can result from mutations, chromosomal abnormalities or adverse environmental effects, and can occur in isolation or in association with numerous syndromes. To date, surgical correction remains the primary treatment for craniosynostosis, but it is associated with complications and with the potential for re-synostosis. There is, therefore, a strong unmet need for new therapies. Here, we provide a comprehensive review of our current understanding of craniosynostosis, including typical craniosynostosis types, their clinical manifestations, cranial suture development, and genetic and environmental causes. Based on studies from animal models, we present a framework for understanding the pathogenesis of craniosynostosis, with an emphasis on the loss of postnatal suture mesenchymal stem cells as an emerging disease-driving mechanism. We evaluate emerging treatment options and highlight the potential of mesenchymal stem cell-based suture regeneration as a therapeutic approach for craniosynostosis.
Keywords: Animal models; Craniosynostosis; Environmental factors; Human genetics; Mesenchymal stem cells; Tissue regeneration.
© 2022. Published by The Company of Biologists Ltd.
Conflict of interest statement
Competing interests The authors declare no competing or financial interests.
Figures
References
-
- Agthe, M., Brügge, J., Garbers, Y., Wandel, M., Kespohl, B., Arnold, P., Flynn, C. M., Lokau, J., Aparicio-Siegmund, S., Bretscher, C.et al. (2018). Mutations in craniosynostosis patients cause defective interleukin-11 receptor maturation and drive craniosynostosis-like disease in mice. Cell Rep. 25, 10-18.e15. 10.1016/j.celrep.2018.09.005 - DOI - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
