

Inhibition of p38 MAPK decreases hyperglycemia-induced nephrin endocytosis and attenuates albuminuria
- PMID: 35451598
- PMCID: PMC9110524
- DOI: 10.1007/s00109-022-02184-5
Inhibition of p38 MAPK decreases hyperglycemia-induced nephrin endocytosis and attenuates albuminuria
Abstract
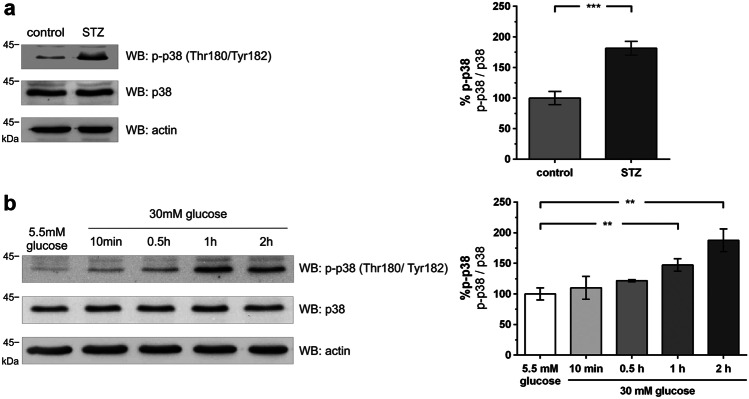
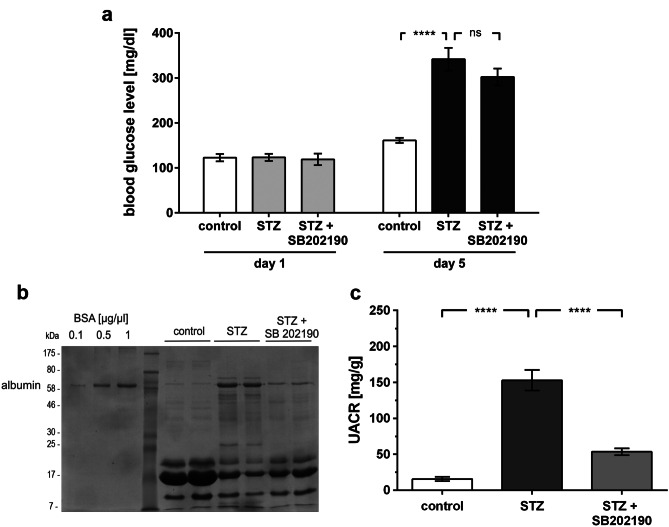
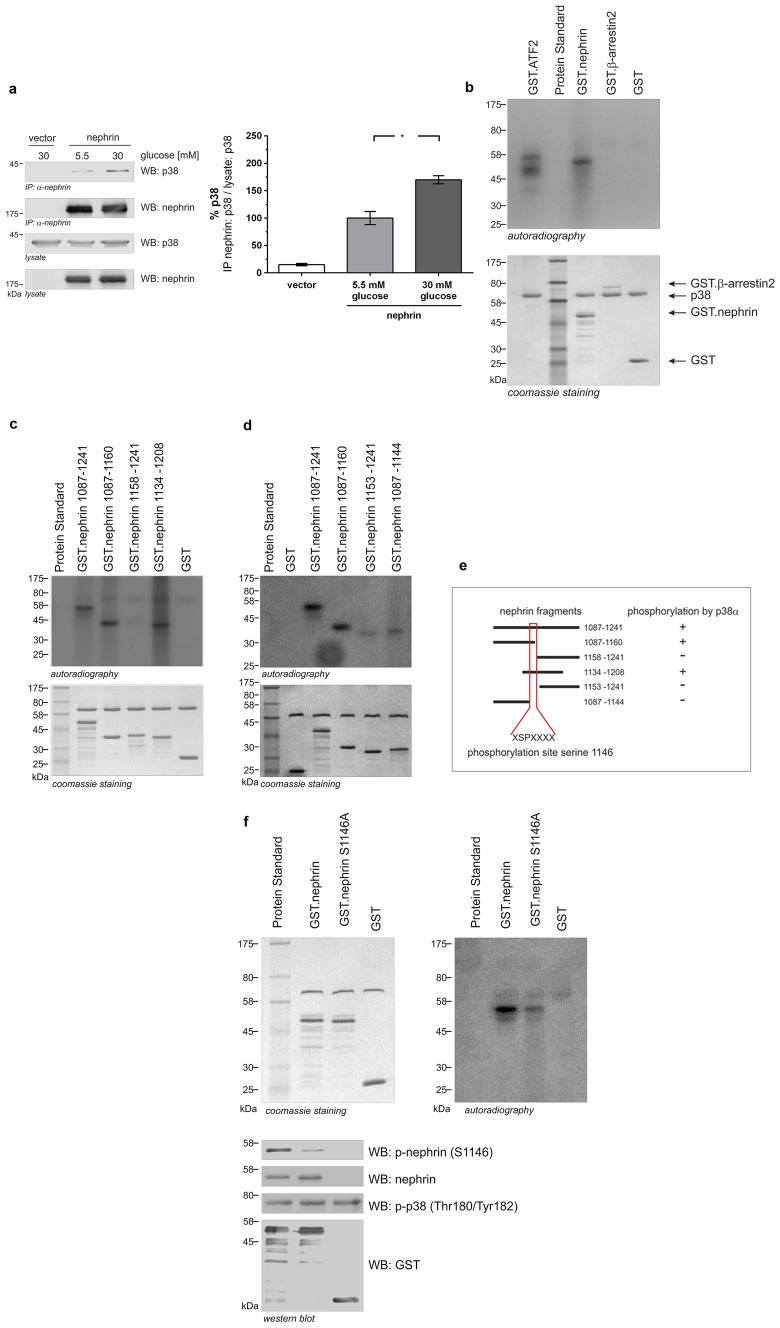
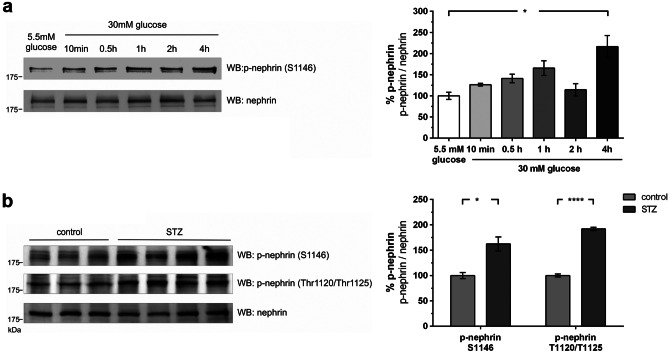
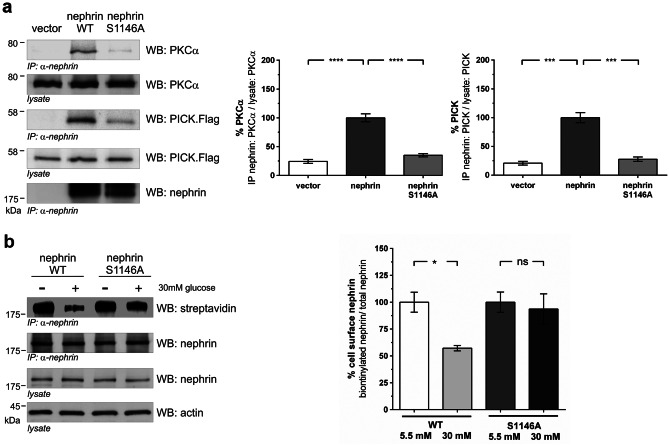
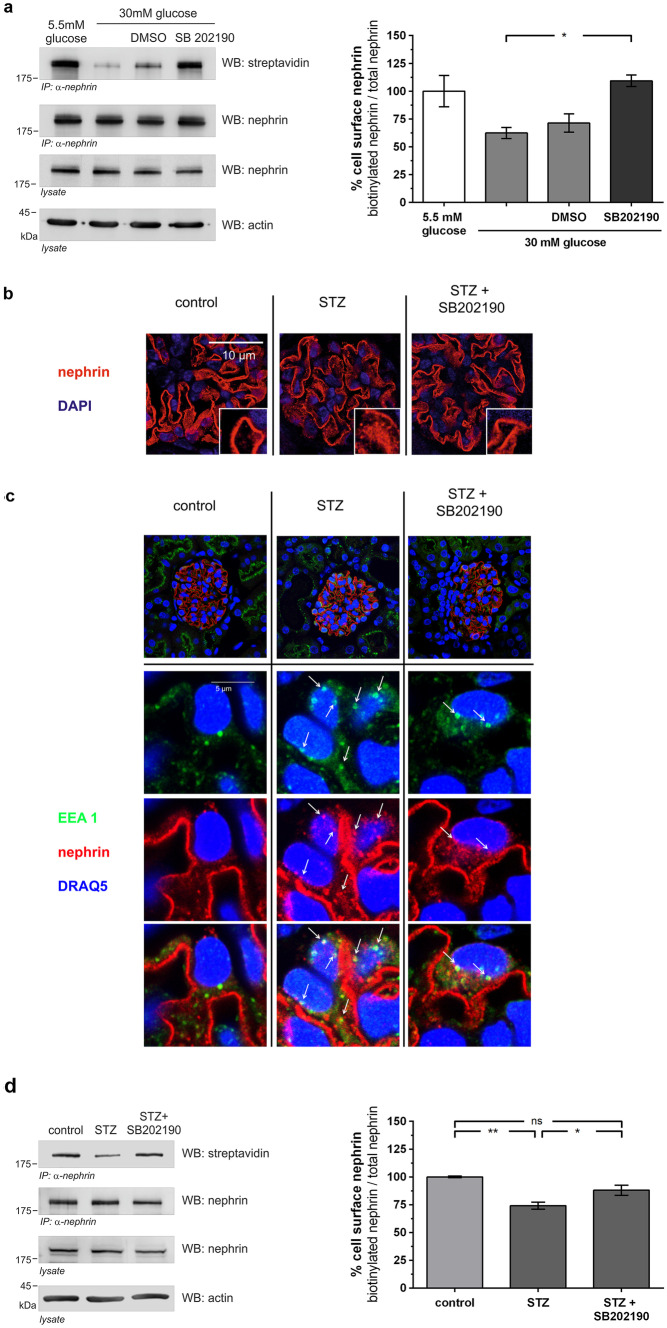
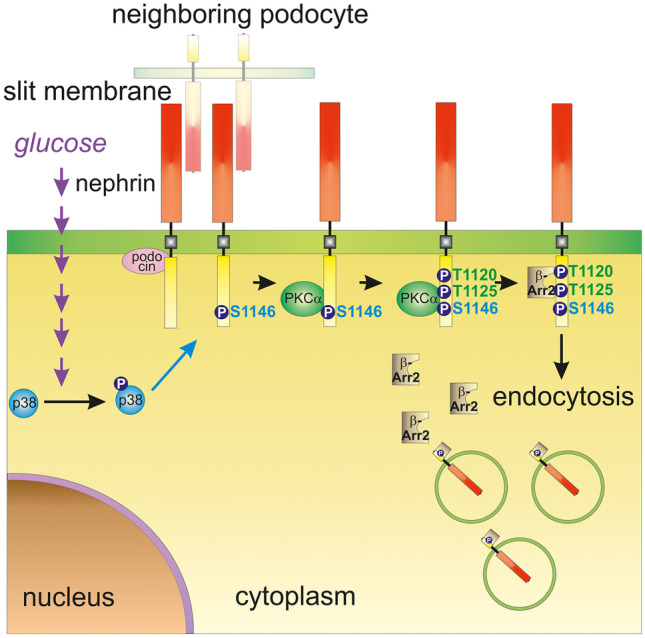
Chronic hyperglycemia, as in diabetes mellitus, may cause glomerular damage with microalbuminuria as an early sign. Noteworthy, even acute hyperglycemia can increase glomerular permeability before structural damage of the glomerular filter can be detected. Despite intensive research, specific antiproteinuric therapy is not available so far. Thus, a deeper understanding of the molecular mechanisms of albuminuria is desirable. P38 MAPK signaling is involved in the development of hyperglycemia-induced albuminuria. However, the mechanism of increased p38 MAPK activity leading to increased permeability and albuminuria remained unclear. Recently, we demonstrated that acute hyperglycemia triggers endocytosis of nephrin, the key molecule of the slit diaphragm, and induces albuminuria. Here, we identify p38 MAPK as a pivotal regulator of hyperglycemia-induced nephrin endocytosis. Activated p38 MAPK phosphorylates the nephrin c-terminus at serine 1146, facilitating the interaction of PKCα with nephrin. PKCα phosphorylates nephrin at threonine residues 1120 and 1125, mediating the binding of β-arrestin2 to nephrin. β-arrestin2 triggers endocytosis of nephrin by coupling it to the endocytic machinery, leading to increased glomerular permeability. Pharmacological inhibition of p38 MAPK preserves nephrin surface expression and significantly attenuates albuminuria. KEY MESSAGES: Acute hyperglycemia triggers endocytosis of nephrin. Activated p38 MAPK phosphorylates the nephrin c-terminus at serine 1146, facilitating the interaction of PKCα with nephrin. PKCα phosphorylates nephrin at threonine residues 1120 and 1125, mediating the binding of β-arrestin2 to nephrin. β-arrestin2 triggers endocytosis of nephrin by coupling it to the endocytic machinery, leading to a leaky glomerular filter. Pharmacological inhibition of p38 MAPK preserves nephrin surface expression and significantly attenuates albuminuria under hyperglycemic conditions.
Keywords: Albuminuria; Diabetes; Endocytosis; Nephrin; Podocyte.
© 2022. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- Chronic Kidney Disease Prognosis C, Matsushita K, van der Velde M, Astor BC, Woodward M, Levey AS, de Jong PE, Coresh J, Gansevoort RT (2010) Association of estimated glomerular filtration rate and albuminuria with all-cause and cardiovascular mortality in general population cohorts: a collaborative meta-analysis. Lancet 375(9731):2073–2081. 10.1016/S0140-6736(10)60674-5 - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
