

Single-EV analysis (sEVA) of mutated proteins allows detection of stage 1 pancreatic cancer
- PMID: 35452280
- PMCID: PMC9032977
- DOI: 10.1126/sciadv.abm3453
Single-EV analysis (sEVA) of mutated proteins allows detection of stage 1 pancreatic cancer
Abstract
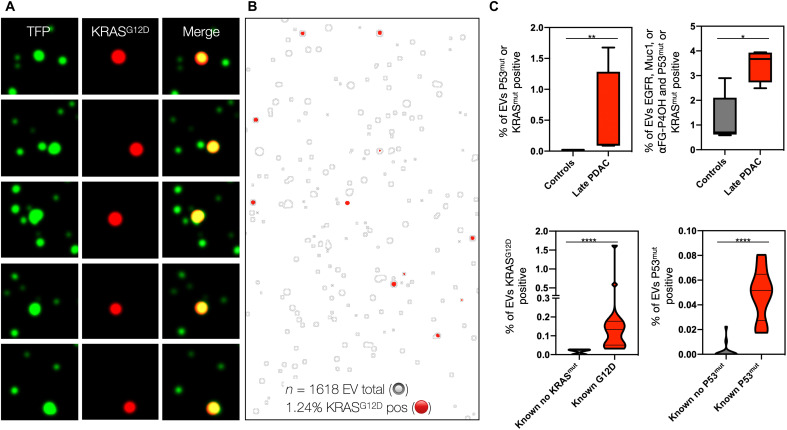
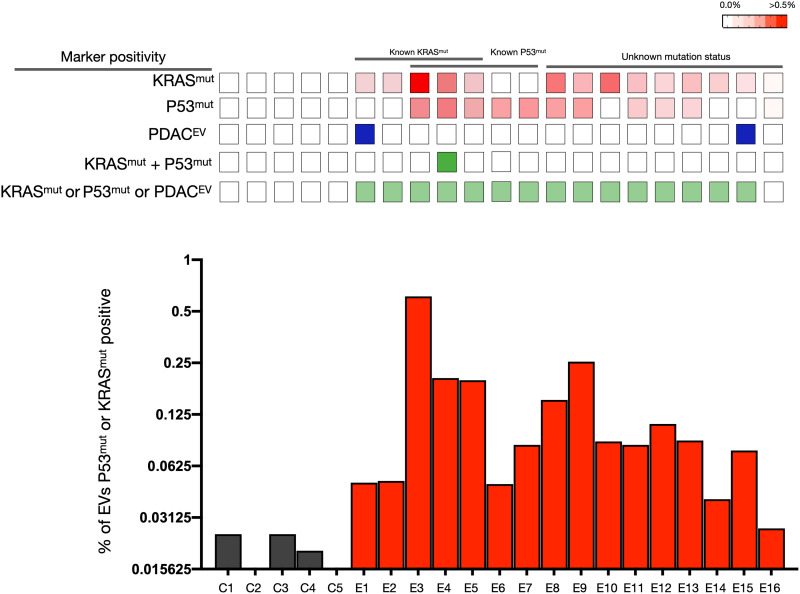
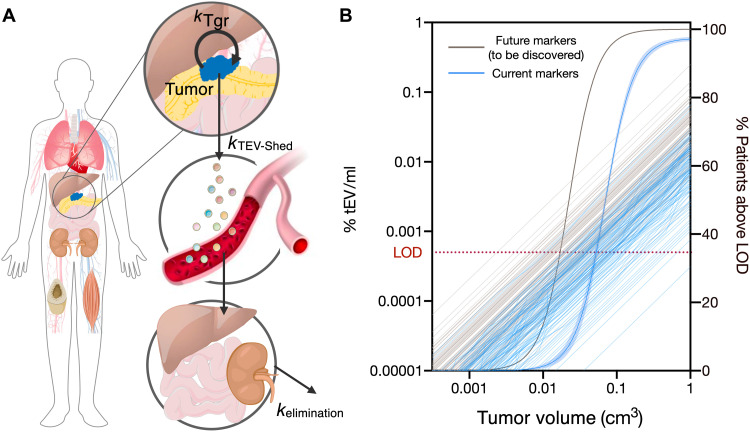
Tumor cell-derived extracellular vesicles (EVs) are being explored as circulating biomarkers, but it is unclear whether bulk measurements will allow early cancer detection. We hypothesized that a single-EV analysis (sEVA) technique could potentially improve diagnostic accuracy. Using pancreatic cancer (PDAC), we analyzed the composition of putative cancer markers in 11 model lines. In parental PDAC cells positive for KRASmut and/or P53mut proteins, only ~40% of EVs were also positive. In a blinded study involving 16 patients with surgically proven stage 1 PDAC, KRASmut and P53mut protein was detectable at much lower levels, generally in <0.1% of vesicles. These vesicles were detectable by the new sEVA approach in 15 of the 16 patients. Using a modeling approach, we estimate that the current PDAC detection limit is at ~0.1-cm3 tumor volume, below clinical imaging capabilities. These findings establish the potential for sEVA for early cancer detection.
Figures
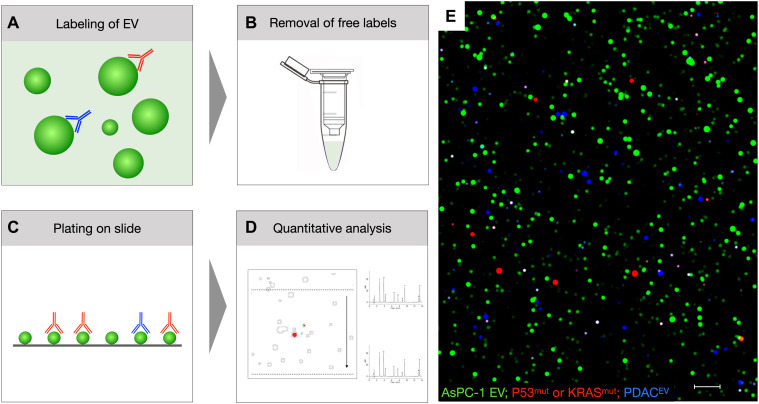
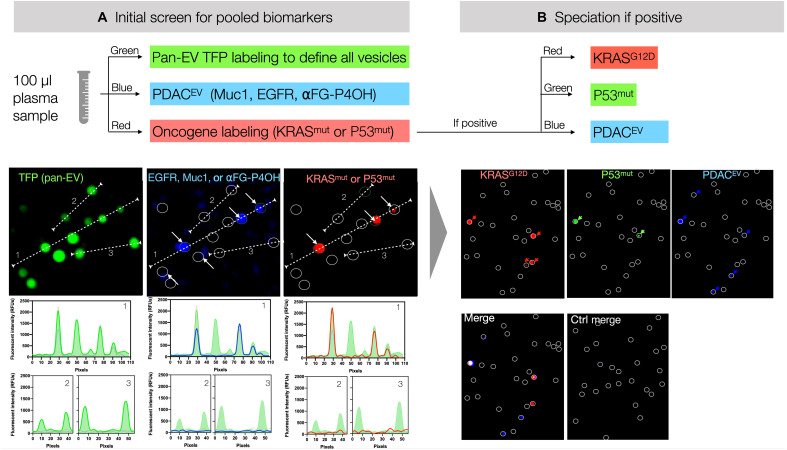
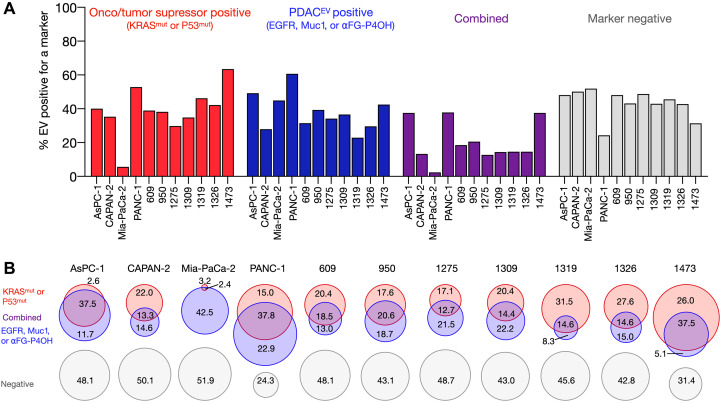
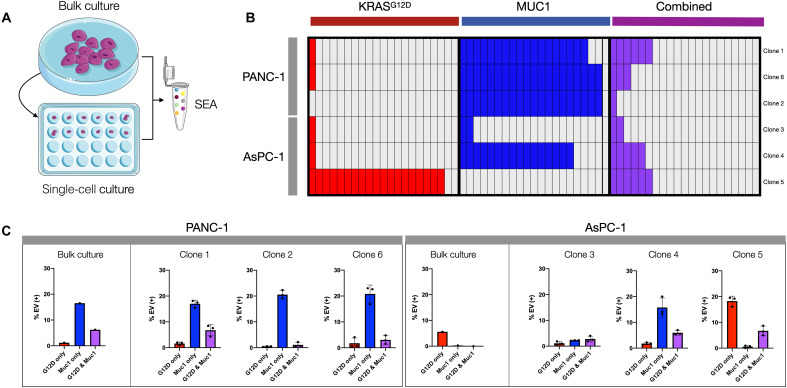
References
-
- Rahib L., Smith B. D., Aizenberg R., Rosenzweig A. B., Fleshman J. M., Matrisian L. M., Projecting cancer incidence and deaths to 2030: The unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 74, 2913–2921 (2014). - PubMed
-
- Ferrone C. R., Marchegiani G., Hong T. S., Ryan D. P., Deshpande V., McDonnell E. I., Sabbatino F., Santos D. D., Allen J. N., Blaszkowsky L. S., Clark J. W., Faris J. E., Goyal L., Kwak E. L., Murphy J. E., Ting D. T., Wo J. Y., Zhu A. X., Warshaw A. L., Lillemoe K. D., Fernández-del Castillo C., Radiological and surgical implications of neoadjuvant treatment with FOLFIRINOX for locally advanced and borderline resectable pancreatic cancer. Ann. Surg. 261, 12–17 (2015). - PMC - PubMed
-
- Mattox A. K., Bettegowda C., Zhou S., Papadopoulos N., Kinzler K. W., Vogelstein B., Applications of liquid biopsies for cancer. Sci. Transl. Med. 11, eaay1984 (2019). - PubMed
-
- Bettegowda C., Sausen M., Leary R. J., Kinde I., Wang Y., Agrawal N., Bartlett B. R., Wang H., Luber B., Alani R. M., Antonarakis E. S., Azad N. S., Bardelli A., Brem H., Cameron J. L., Lee C. C., Fecher L. A., Gallia G. L., Gibbs P., Le D., Giuntoli R. L., Goggins M., Hogarty M. D., Holdhoff M., Hong S. M., Jiao Y., Juhl H. H., Kim J. J., Siravegna G., Laheru D. A., Lauricella C., Lim M., Lipson E. J., Marie S. K., Netto G. J., Oliner K. S., Olivi A., Olsson L., Riggins G. J., Sartore-Bianchi A., Schmidt K., Shih L. M., Oba-Shinjo S. M., Siena S., Theodorescu D., Tie J., Harkins T. T., Veronese S., Wang T. L., Weingart J. D., Wolfgang C. L., Wood L. D., Xing D., Hruban R. H., Wu J., Allen P. J., Schmidt C. M., Choti M. A., Velculescu V. E., Kinzler K. W., Vogelstein B., Papadopoulos N., Diaz L. A., Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci. Transl. Med. 6, 224ra24 (2014). - PMC - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
