

Combining evidence from Mendelian randomization and colocalization: Review and comparison of approaches
- PMID: 35452592
- PMCID: PMC7612737
- DOI: 10.1016/j.ajhg.2022.04.001
Combining evidence from Mendelian randomization and colocalization: Review and comparison of approaches
Abstract
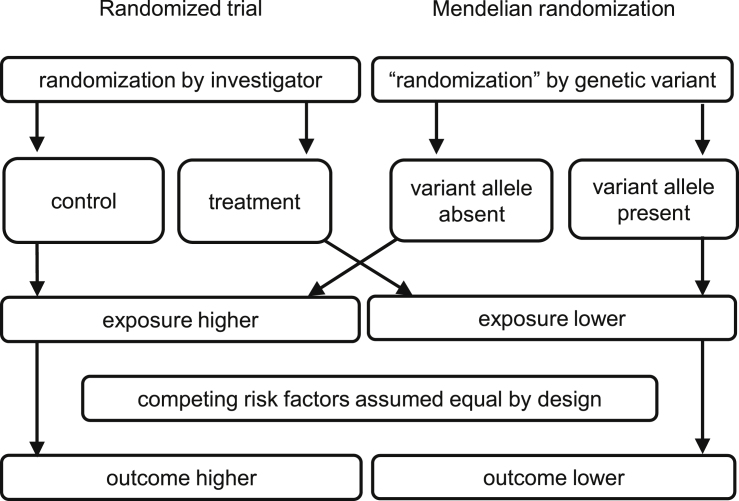
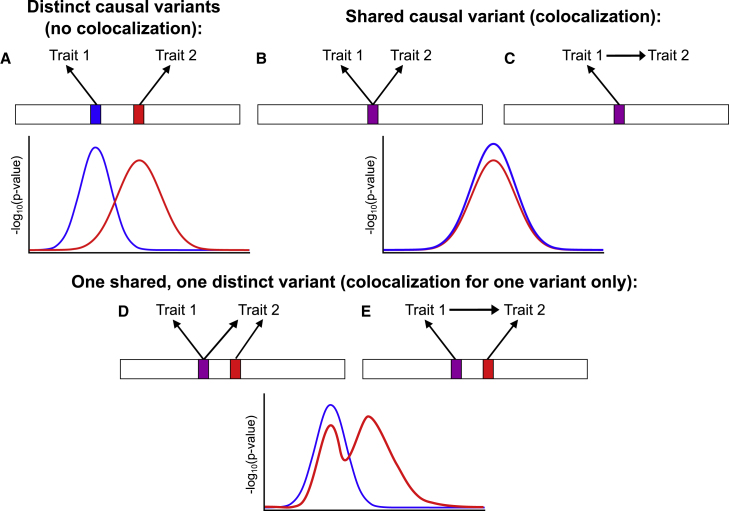
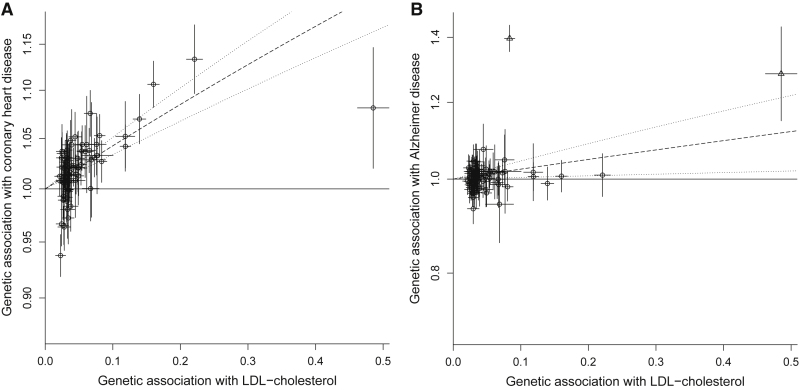
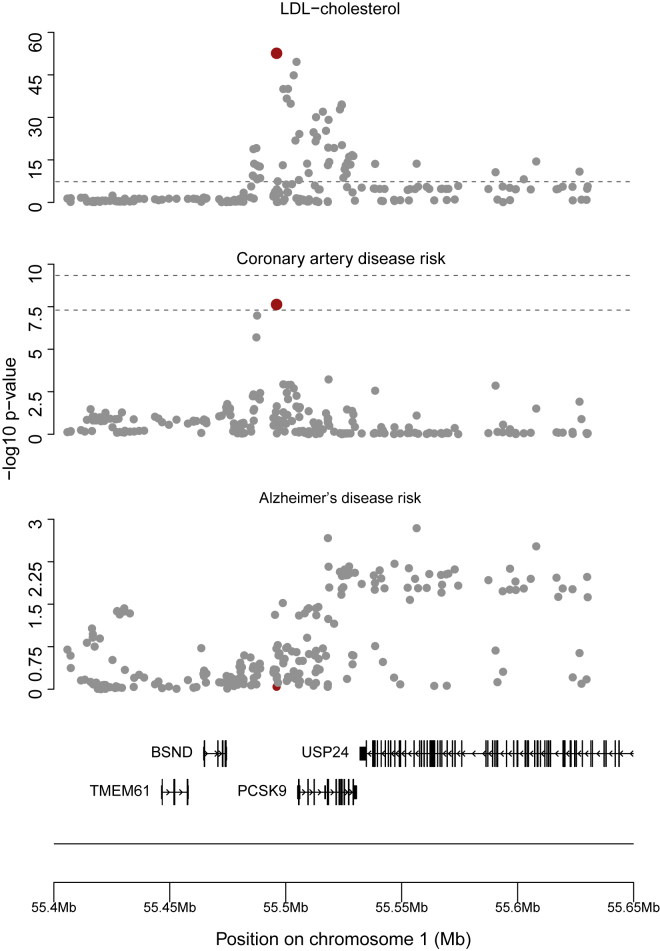
Mendelian randomization and colocalization are two statistical approaches that can be applied to summarized data from genome-wide association studies (GWASs) to understand relationships between traits and diseases. However, despite similarities in scope, they are different in their objectives, implementation, and interpretation, in part because they were developed to serve different scientific communities. Mendelian randomization assesses whether genetic predictors of an exposure are associated with the outcome and interprets an association as evidence that the exposure has a causal effect on the outcome, whereas colocalization assesses whether two traits are affected by the same or distinct causal variants. When considering genetic variants in a single genetic region, both approaches can be performed. While a positive colocalization finding typically implies a non-zero Mendelian randomization estimate, the reverse is not generally true: there are several scenarios which would lead to a non-zero Mendelian randomization estimate but lack evidence for colocalization. These include the existence of distinct but correlated causal variants for the exposure and outcome, which would violate the Mendelian randomization assumptions, and a lack of strong associations with the outcome. As colocalization was developed in the GWAS tradition, typically evidence for colocalization is concluded only when there is strong evidence for associations with both traits. In contrast, a non-zero estimate from Mendelian randomization can be obtained despite only nominally significant genetic associations with the outcome at the locus. In this review, we discuss how the two approaches can provide complementary information on potential therapeutic targets.
Keywords: Causal inference; Genetic epidemiology; phenome-wide association study; post-GWAS investigations; shared heritability.
Copyright © 2022 American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests D.G. is a part-time employee of Novo Nordisk. I.M. and C.W. are wholly or partially funded by a grant from GSK and MSD. The other authors have no relevant conflict of interest to declare.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
