

Regulation of the Homeostatic Unfolded Protein Response in Diabetic Nephropathy
- PMID: 35455399
- PMCID: PMC9030951
- DOI: 10.3390/ph15040401
Regulation of the Homeostatic Unfolded Protein Response in Diabetic Nephropathy
Abstract
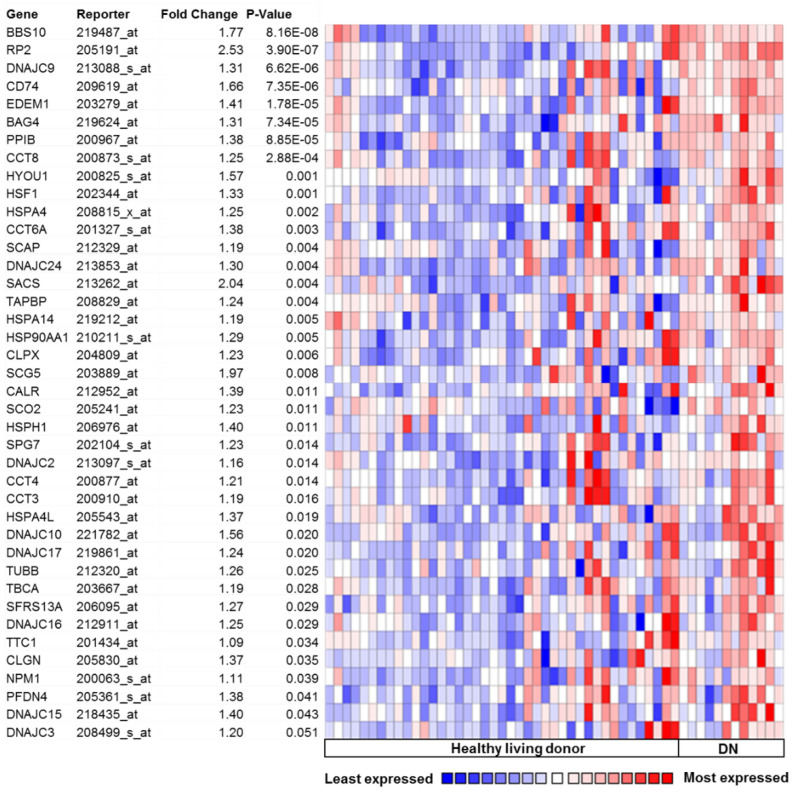
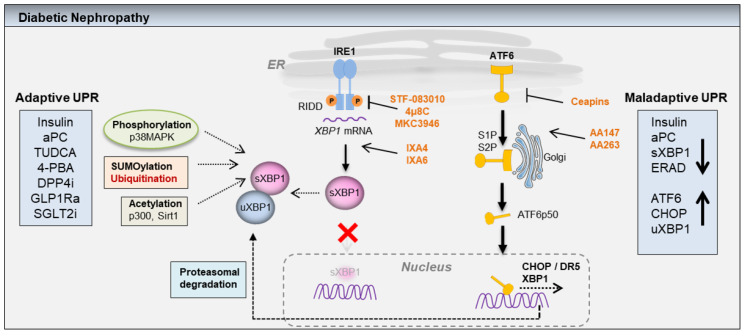
A growing body of scientific evidence indicates that protein homeostasis, also designated as proteostasis, is causatively linked to chronic diabetic nephropathy (DN). Experimental studies have demonstrated that the insulin signaling in podocytes maintain the homeostatic unfolded protein response (UPR). Insulin signaling via the insulin receptor non-canonically activates the spliced X-box binding protein-1 (sXBP1), a highly conserved endoplasmic reticulum (ER) transcription factor, which regulates the expression of genes that control proteostasis. Defective insulin signaling in mouse models of diabetes or the genetic disruption of the insulin signaling pathway in podocytes propagates hyperglycemia induced maladaptive UPR and DN. Insulin resistance in podocytes specifically promotes activating transcription factor 6 (ATF6) dependent pathogenic UPR. Akin to insulin, recent studies have identified that the cytoprotective effect of anticoagulant serine protease-activated protein C (aPC) in DN is mediated by sXBP1. In mouse models of DN, treatment with chemical chaperones that improve protein folding provides an additional benefit on top of currently used ACE inhibitors. Understanding the molecular mechanisms that transmute renal cell specific adaptive responses and that deteriorate renal function in diabetes will enable researchers to develop new therapeutic regimens for DN. Within this review, we focus on the current understanding of homeostatic mechanisms by which UPR is regulated in DN.
Keywords: ATF6; ER stress; XBP1; aPC; diabetic nephropathy; insulin signaling; podocytes; unfolded protein response.
Conflict of interest statement
The authors declare no conflict of interest.
Figures

References
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
