

Pathophysiological Heterogeneity of the BBSOA Neurodevelopmental Syndrome
- PMID: 35455940
- PMCID: PMC9024734
- DOI: 10.3390/cells11081260
Pathophysiological Heterogeneity of the BBSOA Neurodevelopmental Syndrome
Abstract
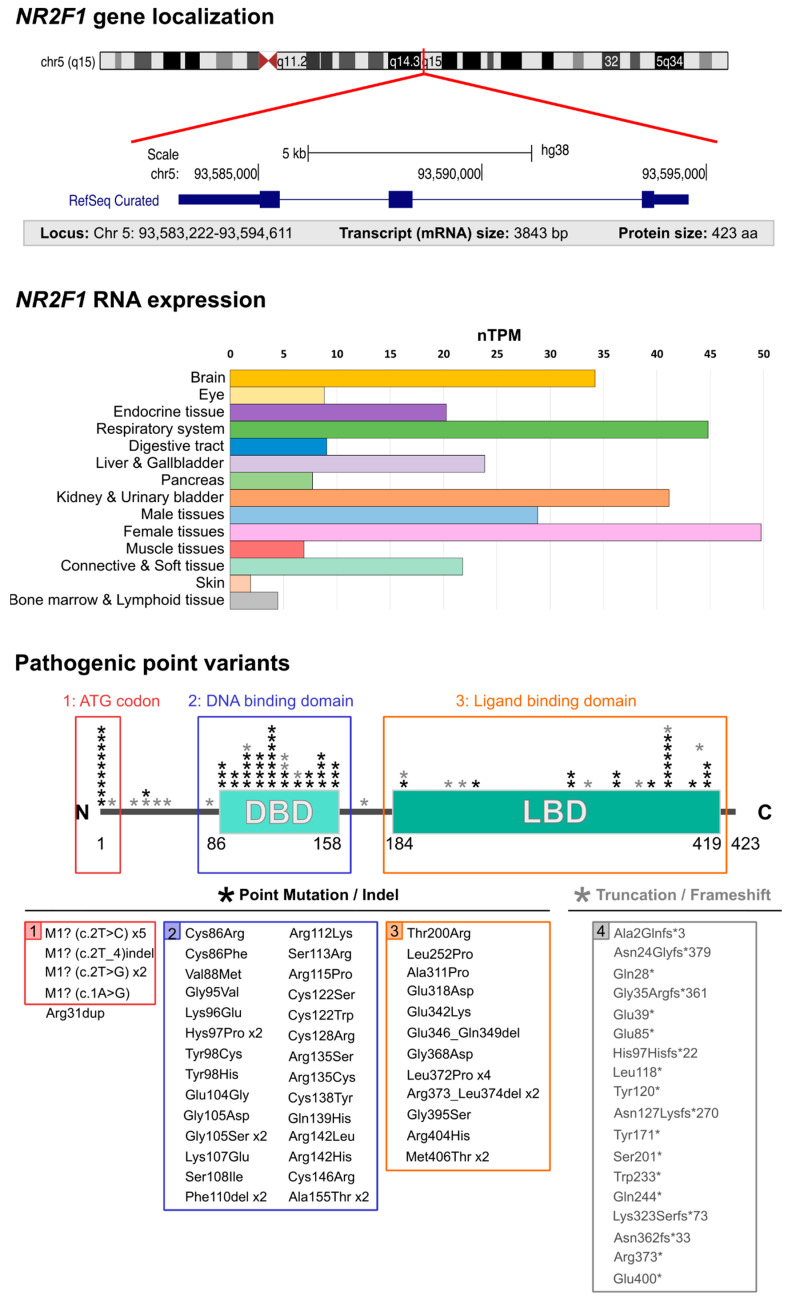
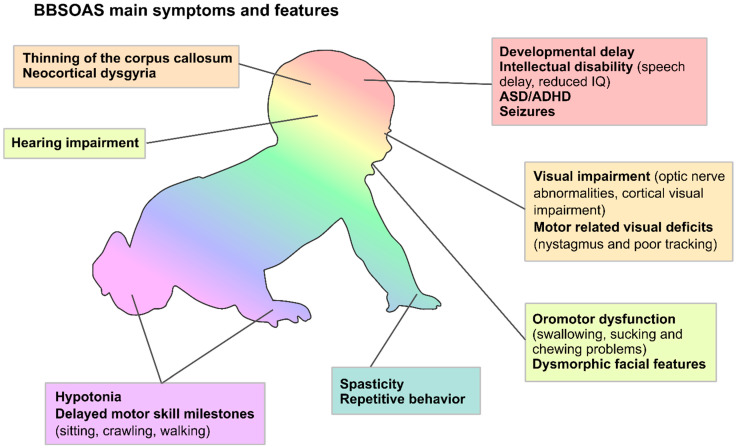
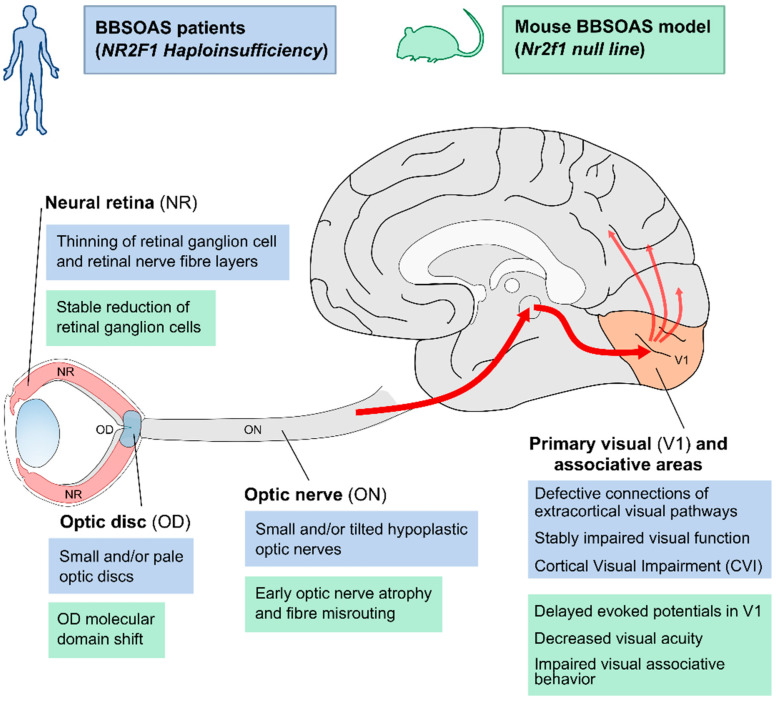
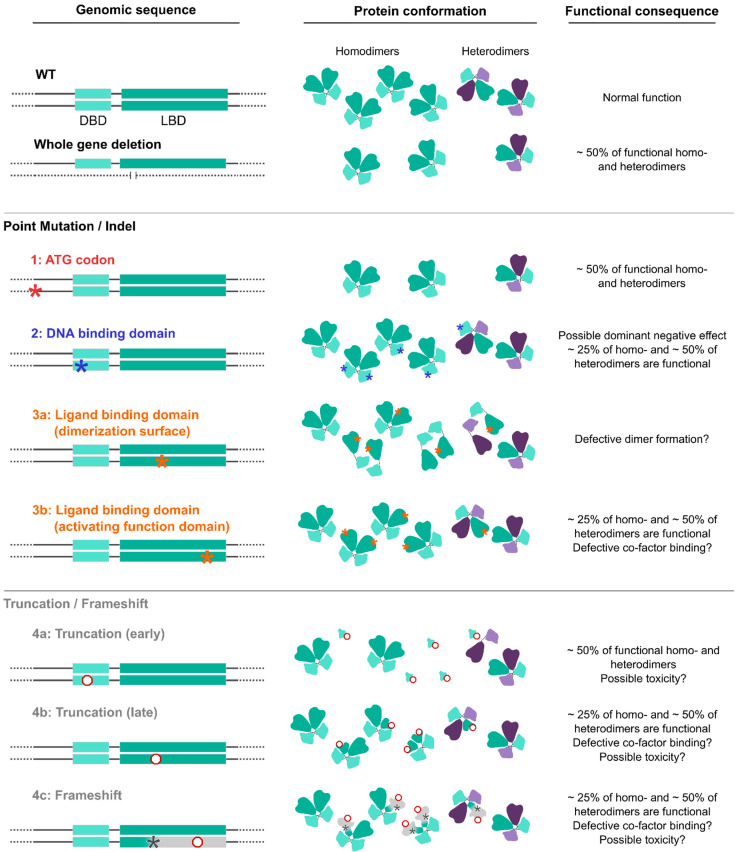
The formation and maturation of the human brain is regulated by highly coordinated developmental events, such as neural cell proliferation, migration and differentiation. Any impairment of these interconnected multi-factorial processes can affect brain structure and function and lead to distinctive neurodevelopmental disorders. Here, we review the pathophysiology of the Bosch-Boonstra-Schaaf Optic Atrophy Syndrome (BBSOAS; OMIM 615722; ORPHA 401777), a recently described monogenic neurodevelopmental syndrome caused by the haploinsufficiency of NR2F1 gene, a key transcriptional regulator of brain development. Although intellectual disability, developmental delay and visual impairment are arguably the most common symptoms affecting BBSOAS patients, multiple additional features are often reported, including epilepsy, autistic traits and hypotonia. The presence of specific symptoms and their variable level of severity might depend on still poorly characterized genotype-phenotype correlations. We begin with an overview of the several mutations of NR2F1 identified to date, then further focuses on the main pathological features of BBSOAS patients, providing evidence-whenever possible-for the existing genotype-phenotype correlations. On the clinical side, we lay out an up-to-date list of clinical examinations and therapeutic interventions recommended for children with BBSOAS. On the experimental side, we describe state-of-the-art in vivo and in vitro studies aiming at deciphering the role of mouse Nr2f1, in physiological conditions and in pathological contexts, underlying the BBSOAS features. Furthermore, by modeling distinct NR2F1 genetic alterations in terms of dimer formation and nuclear receptor binding efficiencies, we attempt to estimate the total amounts of functional NR2F1 acting in developing brain cells in normal and pathological conditions. Finally, using the NR2F1 gene and BBSOAS as a paradigm of monogenic rare neurodevelopmental disorder, we aim to set the path for future explorations of causative links between impaired brain development and the appearance of symptoms in human neurological syndromes.
Keywords: BBSOAS; NR2F1; clinical symptoms; genotype-phenotype correlation; haploinsufficiency; mouse models; neurodevelopmental disorder.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
