

GBA Variants and Parkinson Disease: Mechanisms and Treatments
- PMID: 35455941
- PMCID: PMC9029385
- DOI: 10.3390/cells11081261
GBA Variants and Parkinson Disease: Mechanisms and Treatments
Abstract
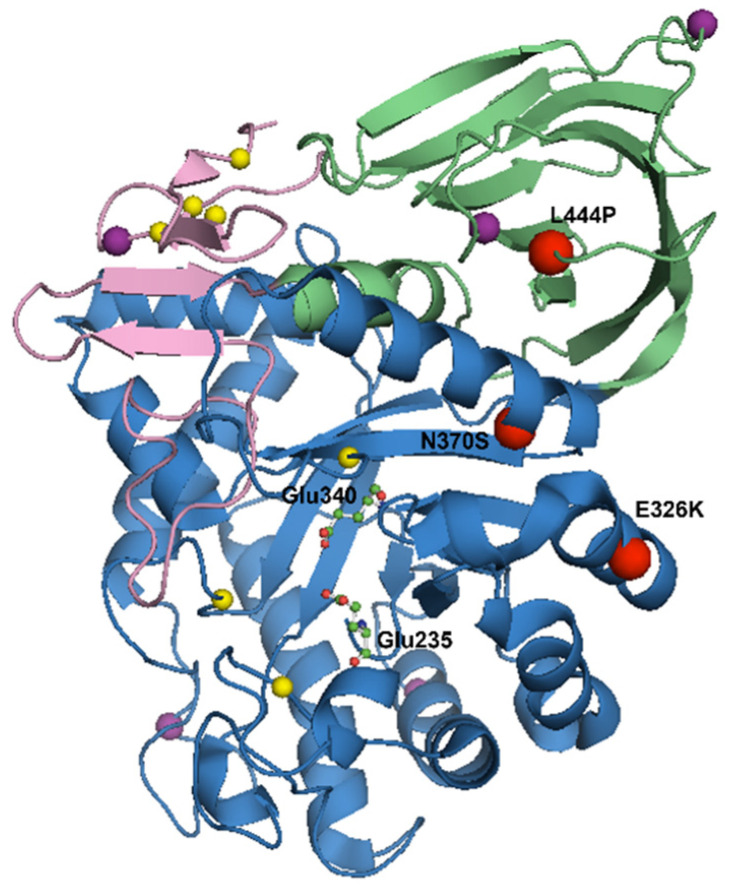
The GBA gene encodes for the lysosomal enzyme glucocerebrosidase (GCase), which maintains glycosphingolipid homeostasis. Approximately 5-15% of PD patients have mutations in the GBA gene, making it numerically the most important genetic risk factor for Parkinson disease (PD). Clinically, GBA-associated PD is identical to sporadic PD, aside from the earlier age at onset (AAO), more frequent cognitive impairment and more rapid progression. Mutations in GBA can be associated with loss- and gain-of-function mechanisms. A key hallmark of PD is the presence of intraneuronal proteinaceous inclusions named Lewy bodies, which are made up primarily of alpha-synuclein. Mutations in the GBA gene may lead to loss of GCase activity and lysosomal dysfunction, which may impair alpha-synuclein metabolism. Models of GCase deficiency demonstrate dysfunction of the autophagic-lysosomal pathway and subsequent accumulation of alpha-synuclein. This dysfunction can also lead to aberrant lipid metabolism, including the accumulation of glycosphingolipids, glucosylceramide and glucosylsphingosine. Certain mutations cause GCase to be misfolded and retained in the endoplasmic reticulum (ER), activating stress responses including the unfolded protein response (UPR), which may contribute to neurodegeneration. In addition to these mechanisms, a GCase deficiency has also been associated with mitochondrial dysfunction and neuroinflammation, which have been implicated in the pathogenesis of PD. This review discusses the pathways associated with GBA-PD and highlights potential treatments which may act to target GCase and prevent neurodegeneration.
Keywords: GBA; Parkinson disease; alpha-synuclein; autophagy; lipids; unfolded protein response.
Conflict of interest statement
The authors declare no conflict of interest.
Figures

References
-
- Turpin J.C., Dubois G., Brice A., Masson M., Nadaud M.C., Boutry J.M., Schram A.W., Tager J.M., Baumann N. Parkinsonian Symptomatology in a Patient with Type I (Adult) Gaucher’s Disease. Springer US; Boston, MA, USA: 1988. pp. 103–105.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
