

Are we there yet? Benchmarking low-coverage nanopore long-read sequencing for the assembling of mitochondrial genomes using the vulnerable silky shark Carcharhinus falciformis
- PMID: 35459089
- PMCID: PMC9027416
- DOI: 10.1186/s12864-022-08482-z
Are we there yet? Benchmarking low-coverage nanopore long-read sequencing for the assembling of mitochondrial genomes using the vulnerable silky shark Carcharhinus falciformis
Abstract
Background: Whole mitochondrial genomes are quickly becoming markers of choice for the exploration of within-species genealogical and among-species phylogenetic relationships. Most often, 'primer walking' or 'long PCR' strategies plus Sanger sequencing or low-pass whole genome sequencing using Illumina short reads are used for the assembling of mitochondrial chromosomes. In this study, we first confirmed that mitochondrial genomes can be sequenced from long reads using nanopore sequencing data exclusively. Next, we examined the accuracy of the long-reads assembled mitochondrial chromosomes when comparing them to a 'gold' standard reference mitochondrial chromosome assembled using Illumina short-reads sequencing.
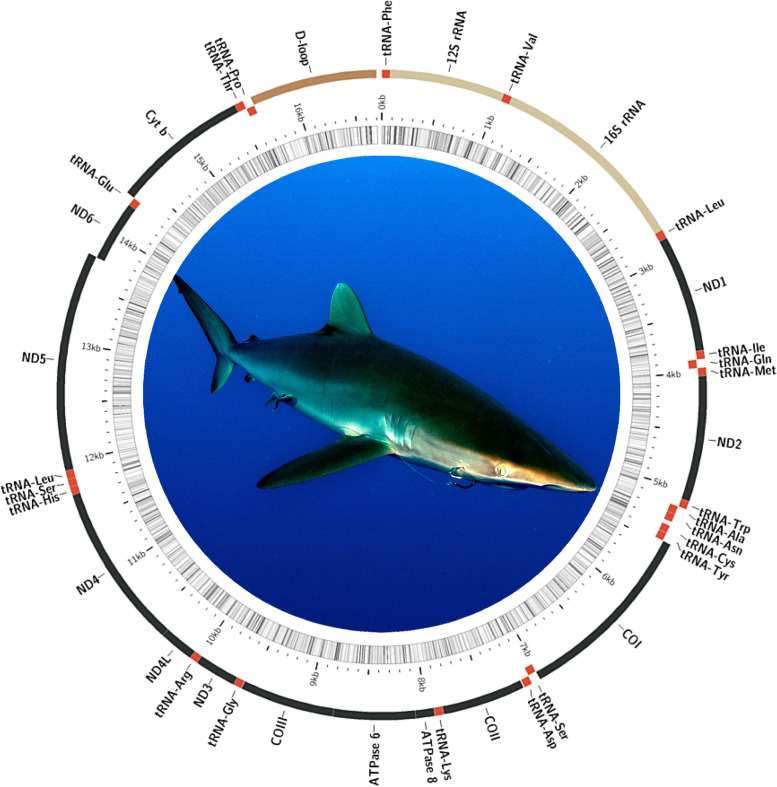
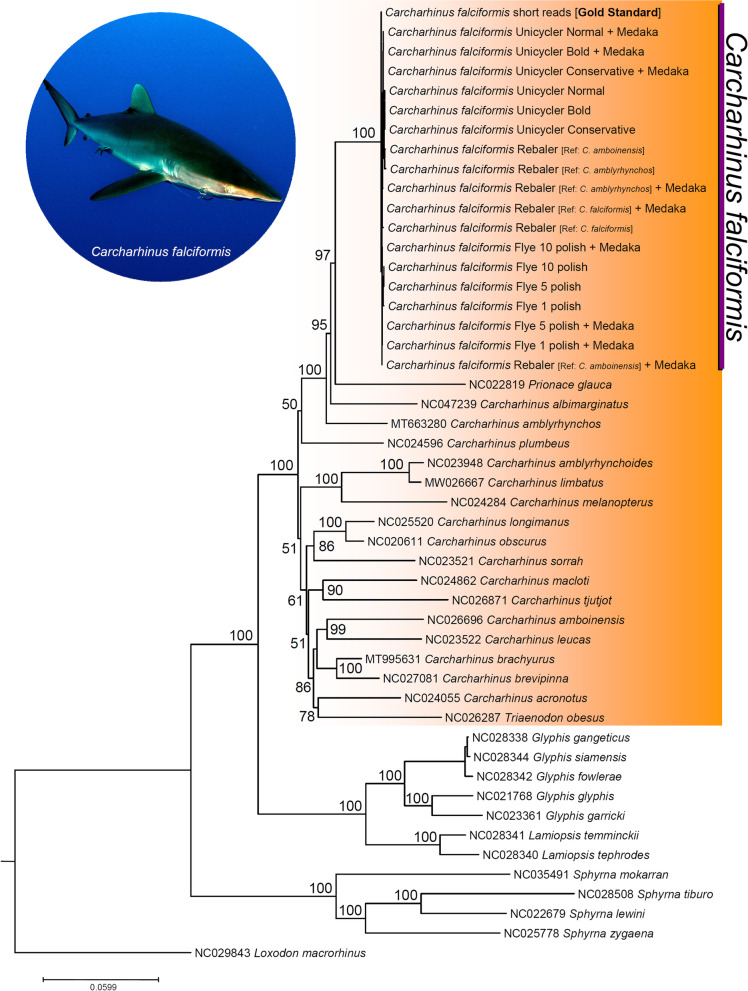
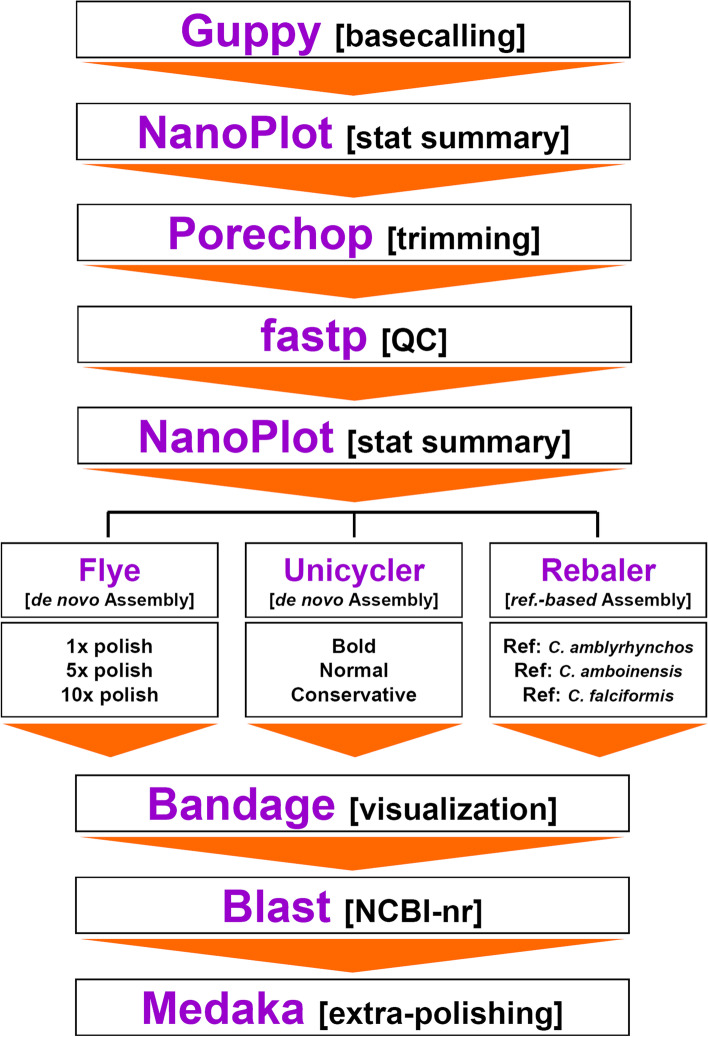
Results: Using a specialized bioinformatics tool, we first produced a short-reads mitochondrial genome assembly for the silky shark C. falciformis with an average base coverage of 9.8x. The complete mitochondrial genome of C. falciformis was 16,705 bp in length and 934 bp shorter than a previously assembled genome (17,639 bp in length) that used bioinformatics tools not specialized for the assembly of mitochondrial chromosomes. Next, low-pass whole genome sequencing using a MinION ONT pocket-sized platform plus customized de-novo and reference-based workflows assembled and circularized a highly accurate mitochondrial genome in the silky shark Carcharhinus falciformis. Indels at the flanks of homopolymer regions explained most of the dissimilarities observed between the 'gold' standard reference mitochondrial genome (assembled using Illumina short reads) and each of the long-reads mitochondrial genome assemblies. Although not completely accurate, mitophylogenomics and barcoding analyses (using entire mitogenomes and the D-Loop/Control Region, respectively) suggest that long-reads assembled mitochondrial genomes are reliable for identifying a sequenced individual, such as C. falciformis, and separating the same individual from others belonging to closely related congeneric species.
Conclusions: This study confirms that mitochondrial genomes can be sequenced from long-reads nanopore sequencing data exclusively. With further development, nanopore technology can be used to quickly test in situ mislabeling in the shark fin fishing industry and thus, improve surveillance protocols, law enforcement, and the regulation of this fishery. This study will also assist with the transferring of high-throughput sequencing technology to middle- and low-income countries so that international scientists can explore population genomics in sharks using inclusive research strategies. Lastly, we recommend assembling mitochondrial genomes using specialized assemblers instead of other assemblers developed for bacterial and/or nuclear genomes.
Keywords: Elasmobranch; Long-read sequencing; Nanopore.
© 2022. The Author(s).
Conflict of interest statement
The author declares no competing interests.
Figures



References
-
- Bernt M, Bleidorn C, Braband A, Dambach J, Donath A, Fritzsch G, et al. A comprehensive analysis of bilaterian mitochondrial genomes and phylogeny. Mol Phylogenet Evol. 2013;69:352–364. - PubMed
-
- Lou RN, Fletcher NK, Wilder AP, Conover DO, Therkildsen NO, Searle JB. Full mitochondrial genome sequences reveal new insights about postglacial expansion and regional phylogeographic structure in the Atlantic silverside (Menidia menidia) Mar Biol. 2018;165(8):124.
-
- Baeza JA, Sepulveda FA, Gonzalez MT. The complete mitochondrial genome and description of a new cryptic species of Benedenia Diesing, 1858 (Monogenea: Capsalidae), a major pathogen infecting the yellowtail kingfish Seriola lalandi Valenciennes in the south-East Pacific. Parasit Vectors. 2019;12:490. - PMC - PubMed
-
- Veldsman WP, Wang Y, Niu J, Baeza JA, Chu KH. Characterization of the complete mitochondrial genome of a coconut crab, Birgus latro (Linnaeus, 1767)(Decapoda: Anomura: Coenobitidae), from Okinawa, Japan. J Crustac Biol. 2020;40:390–400.
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
