

Phylogenetic and phylodynamic approaches to understanding and combating the early SARS-CoV-2 pandemic
- PMID: 35459859
- PMCID: PMC9028907
- DOI: 10.1038/s41576-022-00483-8
Phylogenetic and phylodynamic approaches to understanding and combating the early SARS-CoV-2 pandemic
Abstract
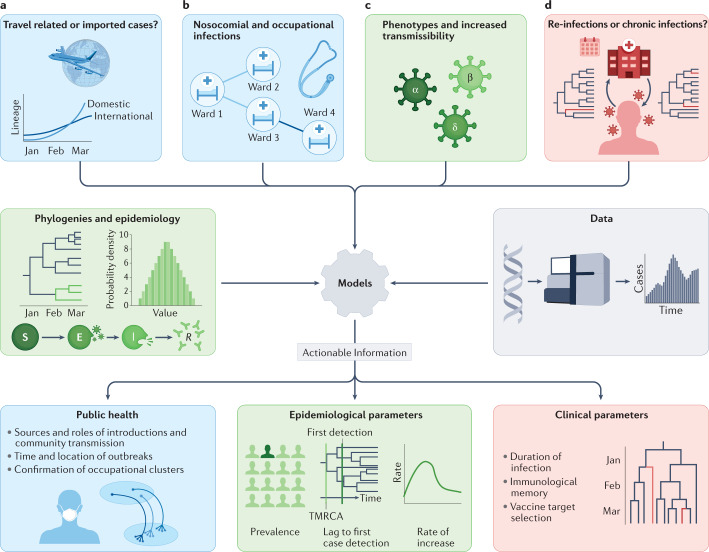
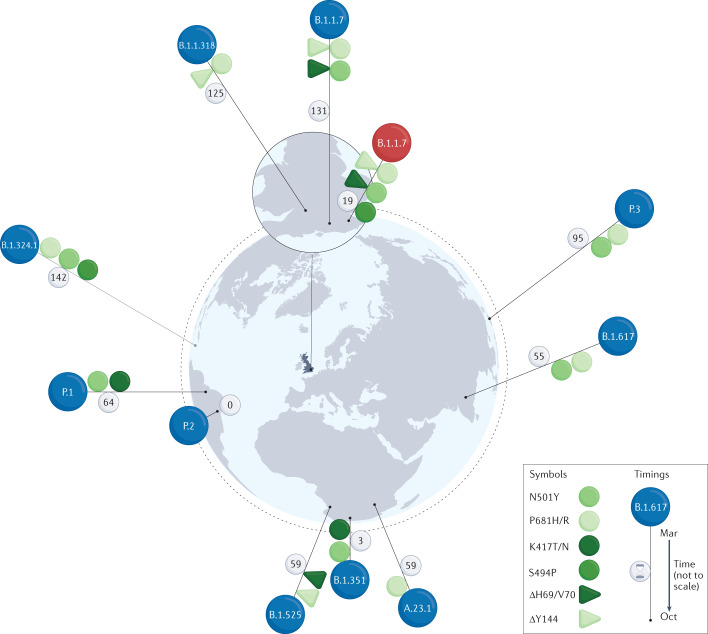
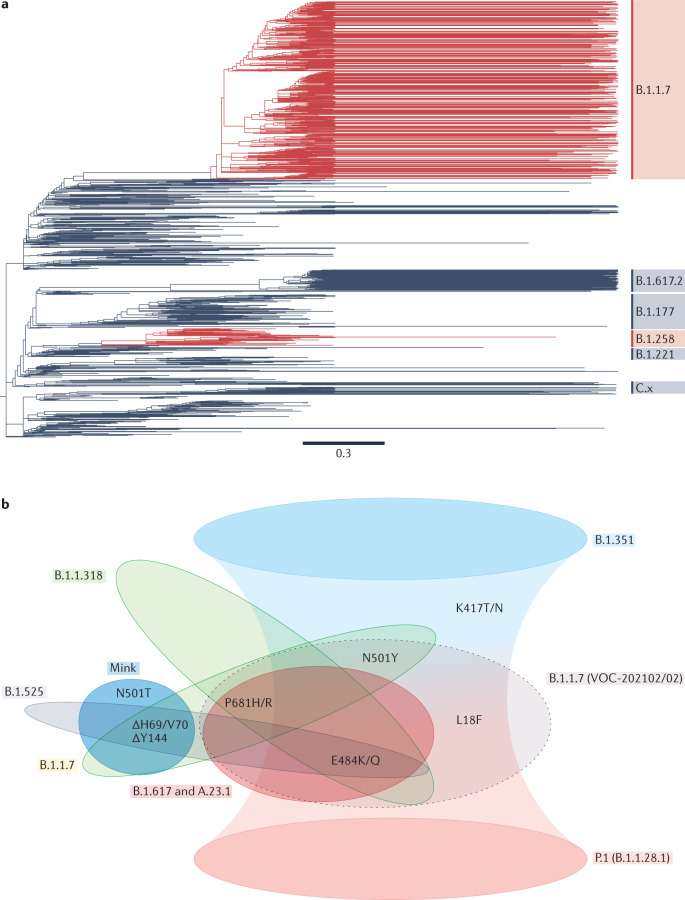
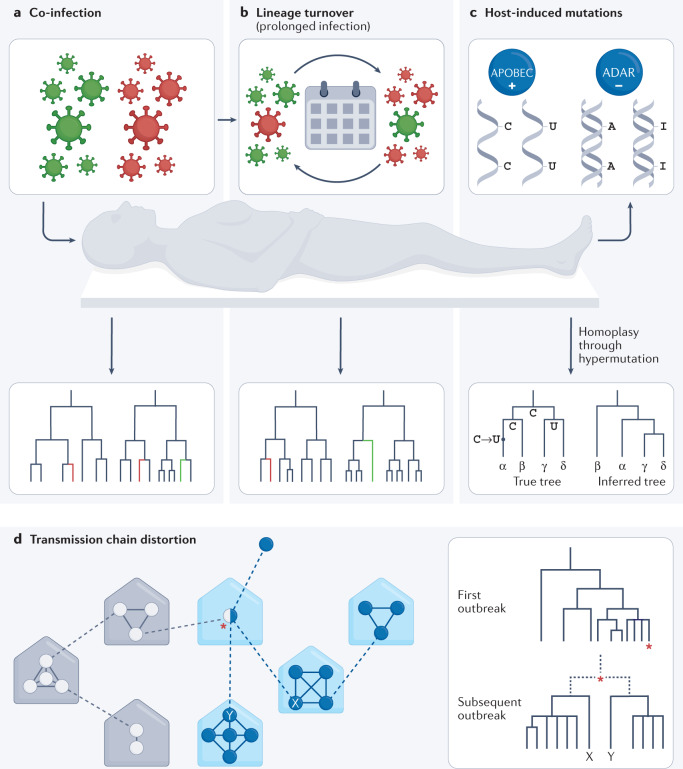
Determining the transmissibility, prevalence and patterns of movement of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infections is central to our understanding of the impact of the pandemic and to the design of effective control strategies. Phylogenies (evolutionary trees) have provided key insights into the international spread of SARS-CoV-2 and enabled investigation of individual outbreaks and transmission chains in specific settings. Phylodynamic approaches combine evolutionary, demographic and epidemiological concepts and have helped track virus genetic changes, identify emerging variants and inform public health strategy. Here, we review and synthesize studies that illustrate how phylogenetic and phylodynamic techniques were applied during the first year of the pandemic, and summarize their contributions to our understanding of SARS-CoV-2 transmission and control.
© 2022. Springer Nature Limited.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
