

Identification of a Rare Variant of c.1777G>A (p.G593S) in the COL1A1 Gene as the Etiology of Recurrent Osteogenesis Imperfecta by Whole-Exome Sequencing
- PMID: 35463886
- PMCID: PMC9028459
- DOI: 10.3389/fped.2022.816090
Identification of a Rare Variant of c.1777G>A (p.G593S) in the COL1A1 Gene as the Etiology of Recurrent Osteogenesis Imperfecta by Whole-Exome Sequencing
Abstract
Background: Osteogenesis imperfecta (OI) is a rare heterogeneous disorder typically featured by fragile bones and susceptibility to fracture. The aim of the present study was to explore the genetic etiology of familial recurrent OI and the genotype-phenotype correlation.
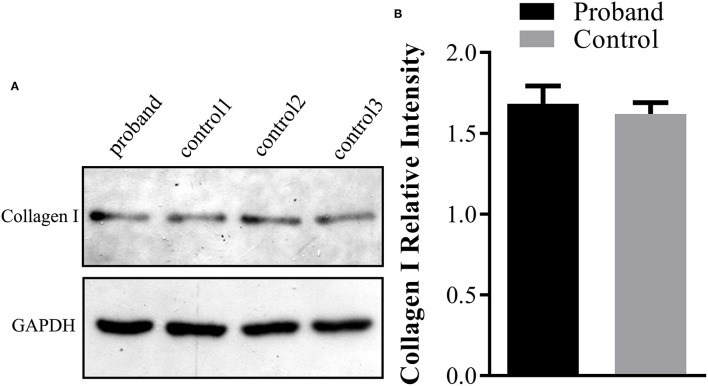
Methods: Karyotyping, chromosomal microarray analysis, and whole-exome sequencing (WES) were performed to determine the genetic etiology of OI in the enrolled family. Western blotting analysis was carried out using the fetal skin tissue for type I collagen production analysis.
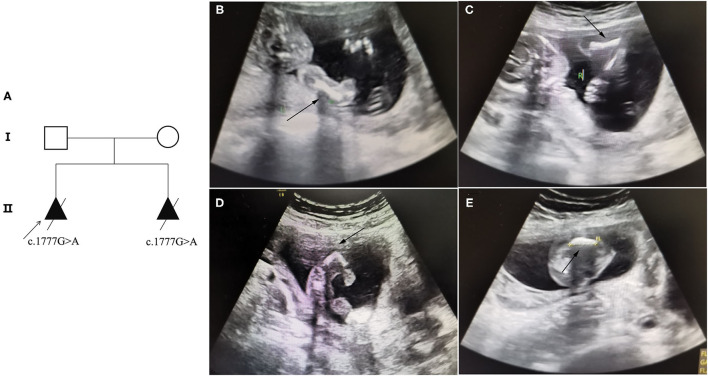
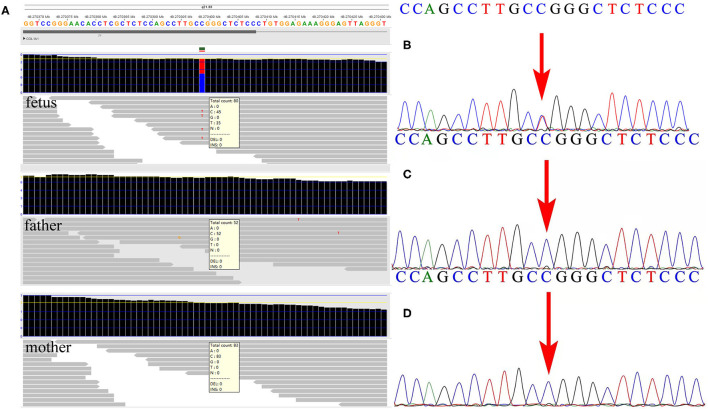
Results: At the first pregnancy, a c.1777G>A mutation in the COL1A1 gene was detected in the fetus who exhibited skeletal dysplasia. In this second pregnancy, severe fetal skeletal dysplasia was also presented without significant chromosomal abnormality detected by karyotype and chromosomal microarray analysis in the fetus. Further WES results demonstrated a de novo missense mutation of c.1777G>A (p.G593S) in the fetus, which was classified as a pathogenic variant according to the ACMG guidelines. The recurrent mutation in the two fetuses hinted at the possible existence of gonadal mosaicism in the parents, while no mutation in the COL1A1 gene was identified in the DNA from the father's sperm. In addition, Western blot results demonstrated no reduced type I procollagen production in the affected fetus compared with the age-matched controls.
Conclusions: To the best of our knowledge, this is the first study that identified a rare variant of c.1777G>A in the COL1A1 gene that led to recurrent OI in the Chinese population. Additionally, we believe that this rare variant of c.1777G>A in the COL1A1 gene will lead to OI type II. The results of the present study further verify the application value of WES in identifying fetuses with ultrasound anomalies.
Keywords: COL1A1; chromosomal microarray analysis; osteogenesis imperfecta; type I collagen; whole-exome sequencing.
Copyright © 2022 Zhuang, Chen, Chen, Luo, Wang, Jiang, Zeng, Xie and Chen.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
Similar articles
-
Etiological identification of recurrent male fatality due to a novel NSDHL gene mutation using trio whole-exome sequencing: A rare case report and literature review.Mol Genet Genomic Med. 2023 Mar;11(3):e2121. doi: 10.1002/mgg3.2121. Epub 2022 Dec 11. Mol Genet Genomic Med. 2023. PMID: 36504312 Free PMC article. Review.
-
Case Report: A novel de novo variant of COL1A1 in fetal genetic osteogenesis imperfecta.Front Endocrinol (Lausanne). 2023 Nov 2;14:1267252. doi: 10.3389/fendo.2023.1267252. eCollection 2023. Front Endocrinol (Lausanne). 2023. PMID: 38027129 Free PMC article.
-
[Analysis of COL1A1 and COL1A2 gene variants in two fetuses with osteogenesis imperfecta].Zhonghua Yi Xue Yi Chuan Xue Za Zhi. 2023 Jul 10;40(7):821-827. doi: 10.3760/cma.j.cn511374-20220722-00486. Zhonghua Yi Xue Yi Chuan Xue Za Zhi. 2023. PMID: 37368383 Chinese.
-
Prenatal Cases Reflect the Complexity of the COL1A1/2 Associated Osteogenesis Imperfecta.Genes (Basel). 2022 Sep 2;13(9):1578. doi: 10.3390/genes13091578. Genes (Basel). 2022. PMID: 36140746 Free PMC article.
-
A novel splicing pathogenic variant in COL1A1 causing osteogenesis imperfecta (OI) type I in a Chinese family.Mol Genet Genomic Med. 2020 Sep;8(9):e1366. doi: 10.1002/mgg3.1366. Epub 2020 Jun 25. Mol Genet Genomic Med. 2020. PMID: 32588564 Free PMC article.
Cited by
-
A de novo PAK1 likely pathogenic variant and a de novo terminal 1q microdeletion in a Chinese girl with global developmental delay, severe intellectual disability, and seizures.BMC Med Genomics. 2023 Jan 9;16(1):3. doi: 10.1186/s12920-023-01433-x. BMC Med Genomics. 2023. PMID: 36624491 Free PMC article.
-
Etiological diagnosis of miscarriage by combining use of chromosomal microarray analysis and whole-exome sequencing.Eur J Med Res. 2025 Jul 1;30(1):528. doi: 10.1186/s40001-025-02709-x. Eur J Med Res. 2025. PMID: 40598400 Free PMC article.
-
Etiological identification of recurrent male fatality due to a novel NSDHL gene mutation using trio whole-exome sequencing: A rare case report and literature review.Mol Genet Genomic Med. 2023 Mar;11(3):e2121. doi: 10.1002/mgg3.2121. Epub 2022 Dec 11. Mol Genet Genomic Med. 2023. PMID: 36504312 Free PMC article. Review.
-
Molecular characterization of a rare TP63 variant associated with split-hand/split-foot malformation 4 and incomplete penetrance: disruption of the p63-Dlx signaling pathway.BMC Genomics. 2025 Feb 5;26(1):113. doi: 10.1186/s12864-025-11297-3. BMC Genomics. 2025. PMID: 39910461 Free PMC article.
-
A De novo Mutation in the COL1A1 Gene Leading to Severe Osteogenesis Imperfecta: Case Report and Review of the Literature.AJP Rep. 2024 Sep 12;14(3):e215-e223. doi: 10.1055/a-2388-3190. eCollection 2024 Jul. AJP Rep. 2024. PMID: 39268228 Free PMC article.
References
-
- Marini JC, Forlino A, Cabral WA, Barnes AM, San Antonio JD, Milgrom S, et al. . Consortium for osteogenesis imperfecta mutations in the helical domain of type I collagen: regions rich in lethal mutations align with collagen binding sites for integrins and proteoglycans. Hum Mutat. (2007) 28:209–21. 10.1002/humu.20429 - DOI - PMC - PubMed
-
- Sillence DO, Rimoin DL, Danks DM. Clinical variability in osteogenesis imperfecta-variable expressivity or genetic heterogeneity. Birth Defects Orig Artic Ser. (1979) 15:113–29. - PubMed
LinkOut - more resources
Full Text Sources
Miscellaneous