

Case Report: A Missense Mutation in Dyskeratosis Congenita 1 Leads to a Benign Form of Dyskeratosis Congenita Syndrome With the Mucocutaneous Triad
- PMID: 35463902
- PMCID: PMC9019361
- DOI: 10.3389/fped.2022.834268
Case Report: A Missense Mutation in Dyskeratosis Congenita 1 Leads to a Benign Form of Dyskeratosis Congenita Syndrome With the Mucocutaneous Triad
Abstract
Background: Dyskeratosis congenita (DC) is a rare inheritable disorder characterized by bone marrow failure and mucocutaneous triad (reticular skin pigmentation, nail dystrophy, and oral leukoplakia). Dyskeratosis congenita 1 (DKC1) is responsible for 4.6% of the DC with an X-linked inheritance pattern. Almost 70 DKC1 variations causing DC have been reported in the Human Gene Mutation Database.
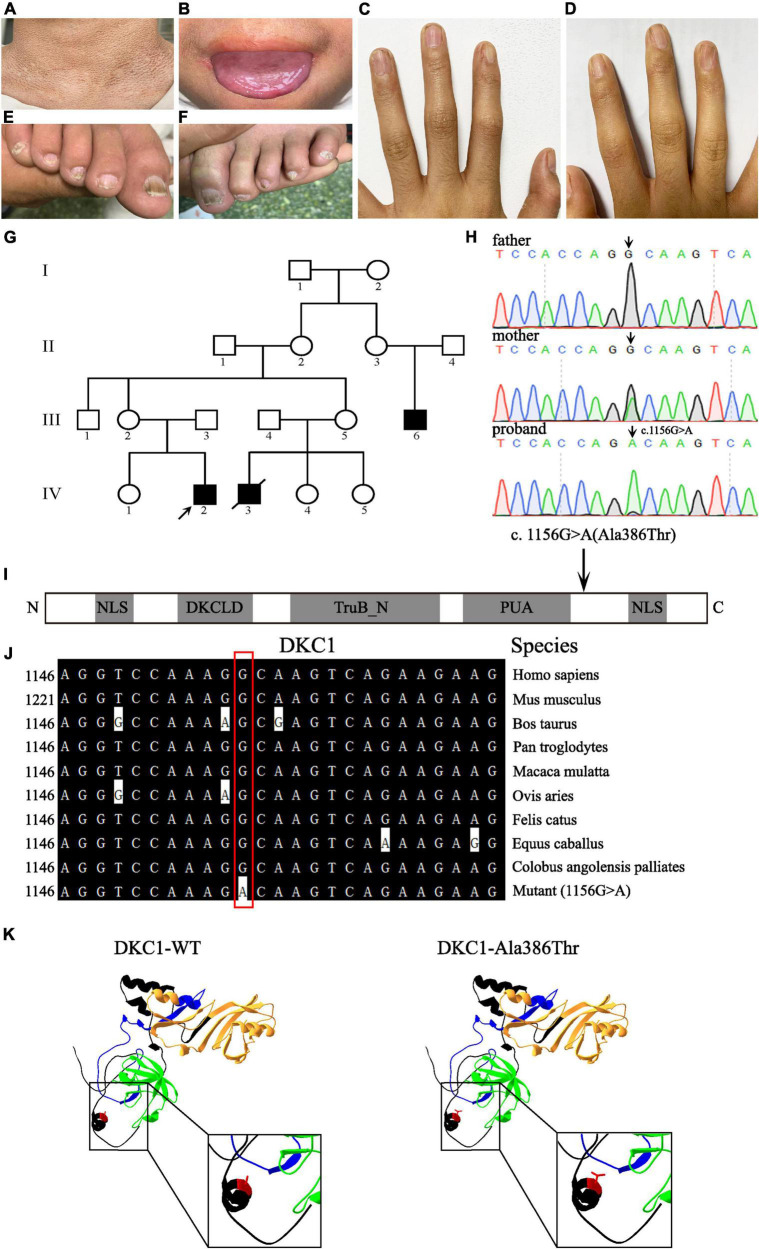
Results: Here we described a 14-year-old boy in a Chinese family with a phenotype of abnormal skin pigmentation on the neck, oral leukoplakia, and nail dysplasia in his hands and feet. Genetic analysis and sequencing revealed hemizygosity for a recurrent missense mutation c.1156G > A (p.Ala386Thr) in DKC1 gene. The heterozygous mutation (c.1156G > A) from his mother and wild-type sequence from his father were obtained in the same site of DKC1. This mutation was determined as disease causing based on silico software, but the pathological phenotypes of the proband were milder than previously reported at this position (HGMDCM060959). Homology modeling revealed that the altered amino acid was located near the PUA domain, which might affect the affinity for RNA binding.
Conclusion: This DKC1 mutation (c.1156G > A, p.Ala386Thr) was first reported in a Chinese family with mucocutaneous triad phenotype. Our study reveals the pathogenesis of DKC1 c.1156G > A mutation to DC with a benign phenotype, which expands the disease variation database, the understanding of genotype-phenotype correlations, and facilitates the clinical diagnosis of DC in China.
Keywords: DKC1; c.1156G > A; dyskeratosis congenita syndrome; missense mutation; p.Ala386Thr.
Copyright © 2022 Wang, Li, Xiong, Zhou, Li and Wu.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
References
Publication types
LinkOut - more resources
Full Text Sources