

Case Report: Diagnostic and Therapeutic Challenges in Severe Mechanobullous Epidermolysis Bullosa Acquisita
- PMID: 35464429
- PMCID: PMC9021387
- DOI: 10.3389/fimmu.2022.883967
Case Report: Diagnostic and Therapeutic Challenges in Severe Mechanobullous Epidermolysis Bullosa Acquisita
Abstract
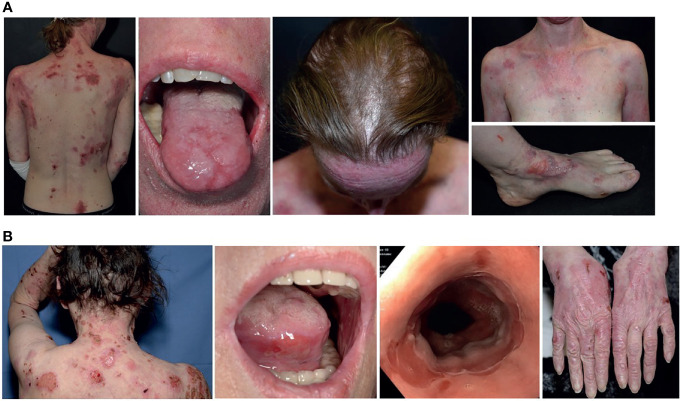
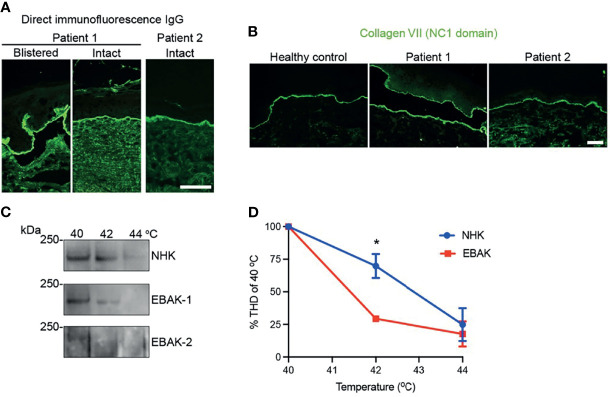
Collagen VII is the main constituent of the anchoring fibrils, important adhesive structures that attach the epidermis to the dermal extracellular matrix. Two disorders are caused by dysfunction of collagen VII, both characterized by skin and mucosa fragility, epidermolysis bullosa acquisita (EBA) and dystrophic epidermolysis bullosa (DEB). EBA and DEB share high clinical similarities with significant difference in patients' age of onset and pathogenesis. Our patients presented with severe and recalcitrant mechanobullous EBA with characteristic DIF, IIF and ELISA diagnostics. But in both women recessive COL7A1 variants were also found, in a monoallelic state. Collagen VII from EBA keratinocytes of our cases was significantly more vulnerable to proteolytic degradation than control keratinocytes, hinting that the heterozygous pathogenic variants were sufficient to destabilize the molecule in vitro. Thus, even if the amount and functionality of mutant and normal type VII collagen polypeptides is sufficient to assure dermal-epidermal adhesion in healthy individuals, the functionally-impaired proteins are probably more prone to development of autoantibodies against them. Our work suggests that testing for COL7A1 genetic variants should be considered in patients with EBA, which either have a patient history hinting towards underlying dystrophic epidermolysis bullosa or pose therapeutic challenges.
Keywords: COL7A1; collagen VII; immunoglobulin; rituximab; skin blistering; skin fragility.
Copyright © 2022 Schauer, Nyström, Kunz, Hübner, Scholl, Athanasiou, Alter, Fischer, Has and Kiritsi.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
Similar articles
-
Epidermolysis Bullosa (EB) Acquisita in an Adult Patient with Previously Unrecognized Mild Dystrophic EB and Biallelic COL7A1 Mutations.Acta Derm Venereol. 2018 Apr 16;98(4):411-415. doi: 10.2340/00015555-2851. Acta Derm Venereol. 2018. PMID: 29182795
-
Autoimmunity to type VII collagen: epidermolysis bullosa acquisita.Curr Dir Autoimmun. 2008;10:195-205. doi: 10.1159/000131455. Curr Dir Autoimmun. 2008. PMID: 18460887 Review.
-
Autoimmunity to type VII collagen: epidermolysis bullosa acquisita.Clin Rev Allergy Immunol. 2007 Oct;33(1-2):78-84. doi: 10.1007/s12016-007-0027-6. Clin Rev Allergy Immunol. 2007. PMID: 18058258 Review.
-
Epidermolysis bullosa acquisita.J Eur Acad Dermatol Venereol. 2013 Oct;27(10):1204-13. doi: 10.1111/jdv.12096. Epub 2013 Feb 1. J Eur Acad Dermatol Venereol. 2013. PMID: 23368767 Review.
-
Epidermal aspects of type VII collagen: Implications for dystrophic epidermolysis bullosa and epidermolysis bullosa acquisita.J Dermatol. 2018 May;45(5):515-521. doi: 10.1111/1346-8138.14222. Epub 2018 Jan 20. J Dermatol. 2018. PMID: 29352483 Review.
Cited by
-
Occurrence of autoantibodies against skin proteins in patients with hereditary epidermolysis bullosa predisposes to development of autoimmune blistering disease.Front Immunol. 2022 Jul 25;13:945176. doi: 10.3389/fimmu.2022.945176. eCollection 2022. Front Immunol. 2022. PMID: 35958577 Free PMC article.
-
Rituximab in the Treatment of Epidermolysis Bullosa Acquisita: A Systematic Review.J Clin Aesthet Dermatol. 2024 Jul;17(7):24-36. J Clin Aesthet Dermatol. 2024. PMID: 39006807 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources