

Multi-site implementation of whole genome sequencing for hospital infection control: A prospective genomic epidemiological analysis
- PMID: 35465046
- PMCID: PMC9019234
- DOI: 10.1016/j.lanwpc.2022.100446
Multi-site implementation of whole genome sequencing for hospital infection control: A prospective genomic epidemiological analysis
Abstract
Background: Current microbiological methods lack the resolution to accurately identify multidrug-resistant organism (MDRO) transmission, however, whole genome sequencing can identify highly-related patient isolates providing opportunities for precision infection control interventions. We investigated the feasibility and potential impact of a prospective multi-centre genomics workflow for hospital infection control.
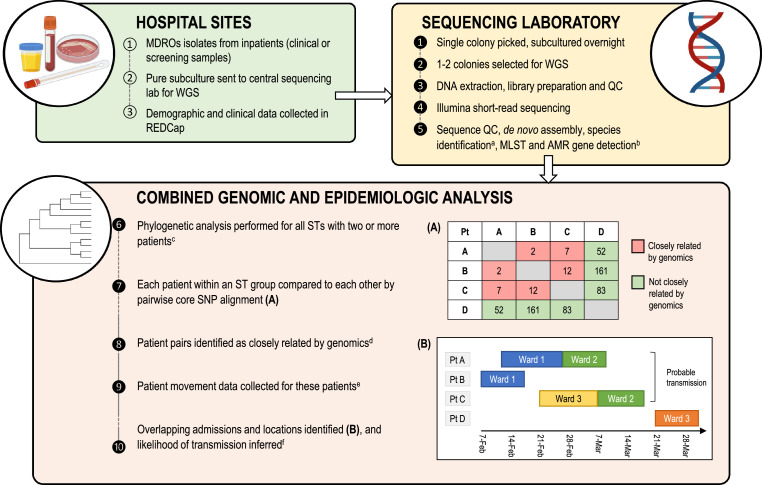
Methods: We conducted a prospective genomics implementation study across eight Australian hospitals over 15 months (2017,2018), collecting all clinical and screening isolates from inpatients with vanA VRE, MRSA, ESBL Escherichia coli (ESBL-Ec), or ESBL Klebsiella pneumoniae (ESBL-Kp). Genomic and epidemiologic data were integrated to assess MDRO transmission.
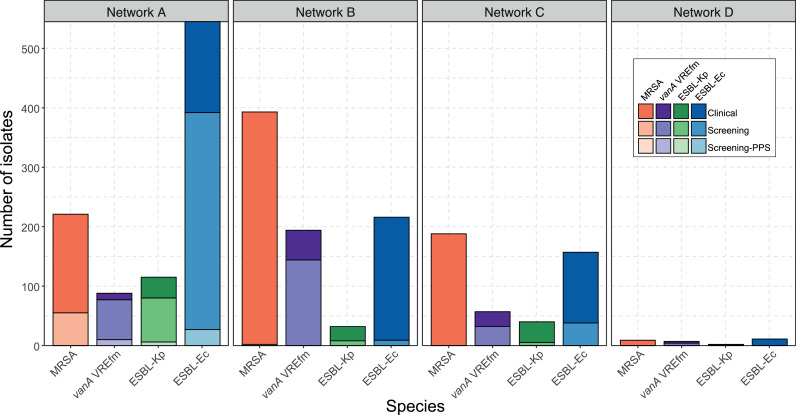
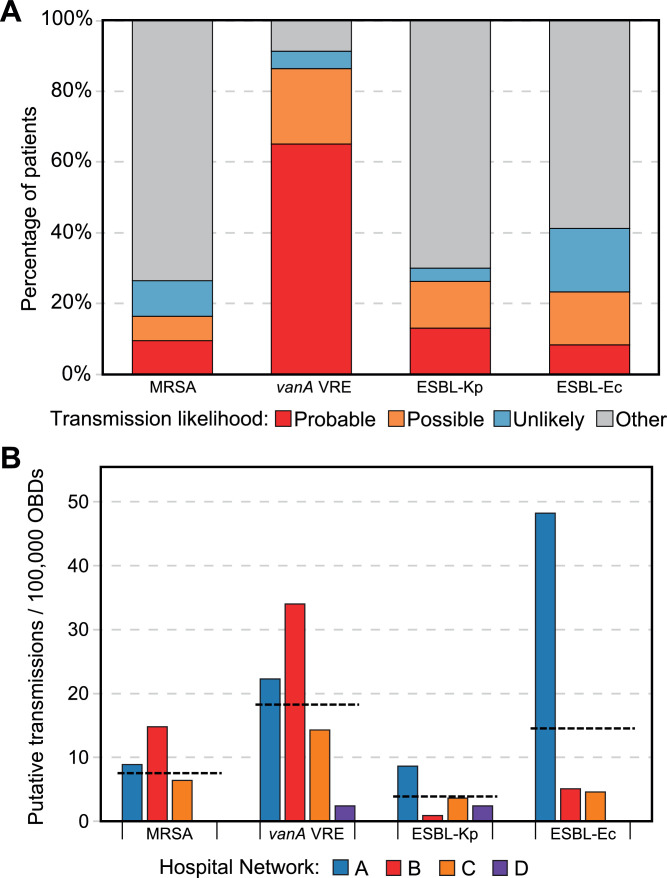
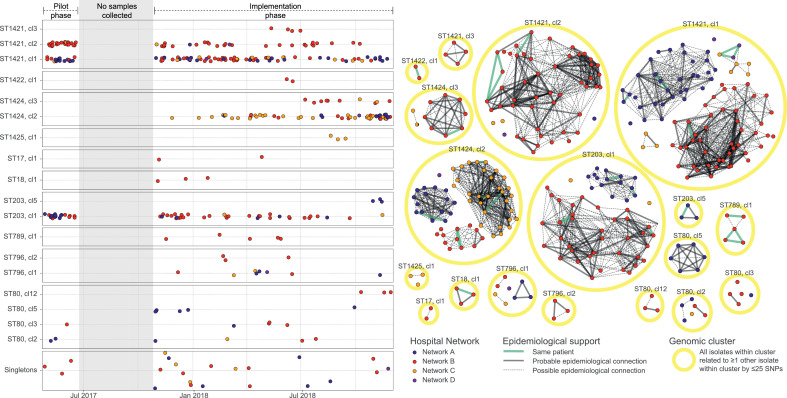
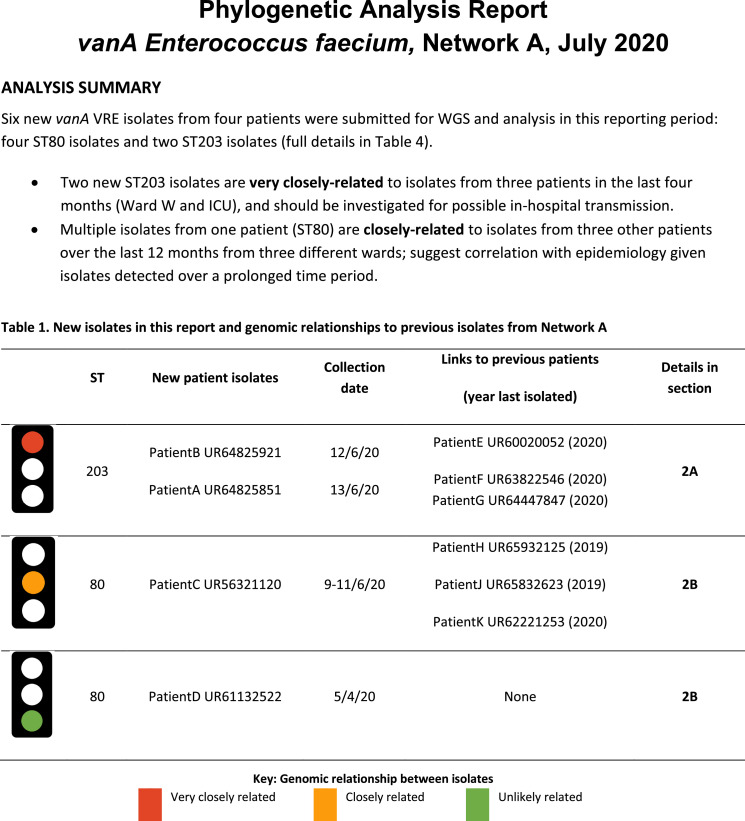
Findings: In total, 2275 isolates were included from 1970 patients, predominantly ESBL-Ec (40·8%) followed by MRSA (35·6%), vanA VRE (15·2%), and ESBL-Kp (8·3%).Overall, hospital and genomic epidemiology showed 607 patients (30·8%) acquired their MDRO in hospital, including the majority of vanA VRE (266 patients, 86·4%), with lower proportions of ESBL-Ec (186 patients, 23·0%), ESBL-Kp (42 patients, 26·3%), and MRSA (113 patients, 16·3%). Complex patient movements meant the majority of MDRO transmissions would remain undetected without genomic data.The genomics implementation had major impacts, identifying unexpected MDRO transmissions prompting new infection control interventions, and contributing to vanA VRE becoming a notifiable condition. We identified barriers to implementation and recommend strategies for mitigation.
Interpretation: Implementation of a multi-centre genomics-informed infection control workflow is feasible and identifies many unrecognised MDRO transmissions. This provides critical opportunities for interventions to improve patient safety in hospitals.
Funding: Melbourne Genomics Health Alliance (supported by State Government of Victoria, Australia), and National Health and Medical Research Council (Australia).
Keywords: Antimicrobial resistance; ESBL-Ec, Extended-spectrum beta-lactamase Escherichia coli; ESBL-Kp, Extended-spectrum beta-lactamase Klebsiella pneumoniae; Hospital epidemiology; Infection prevention and control; MDRO, Multidrug-resistant organism; MRSA, Methicillin-resistant Staphylococcus aureus; VRE, Vancomycin-resistant Enterococcus; WGS, Whole genome sequencing; Whole genome sequencing.
© 2022 The Authors.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures
References
-
- World Health Organization . WHO Press; Geneva, Switzerland: 2014. Antimicrobial Resistance: Global Report on Surveillance.
-
- Kritsotakis E., Kontopidou F., Astrinaki E., Roumbelaki M., Ioannidou E., Gikas A. Prevalence, incidence burden, and clinical impact of healthcare-associated infections and antimicrobial resistance: a national prevalent cohort study in acute care hospitals in Greece. Infect Drug Resist. 2017;10:317–328. - PMC - PubMed
-
- Popovich K.J., Snitkin E.S. Whole genome sequencing-implications for infection prevention and outbreak investigations. Curr Infect Dis Rep. 2017;19(4):15. - PubMed
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
