

Duchenne muscular dystrophy trajectory in R-DMDdel52 preclinical rat model identifies COMP as biomarker of fibrosis
- PMID: 35468843
- PMCID: PMC9036715
- DOI: 10.1186/s40478-022-01355-2
Duchenne muscular dystrophy trajectory in R-DMDdel52 preclinical rat model identifies COMP as biomarker of fibrosis
Abstract
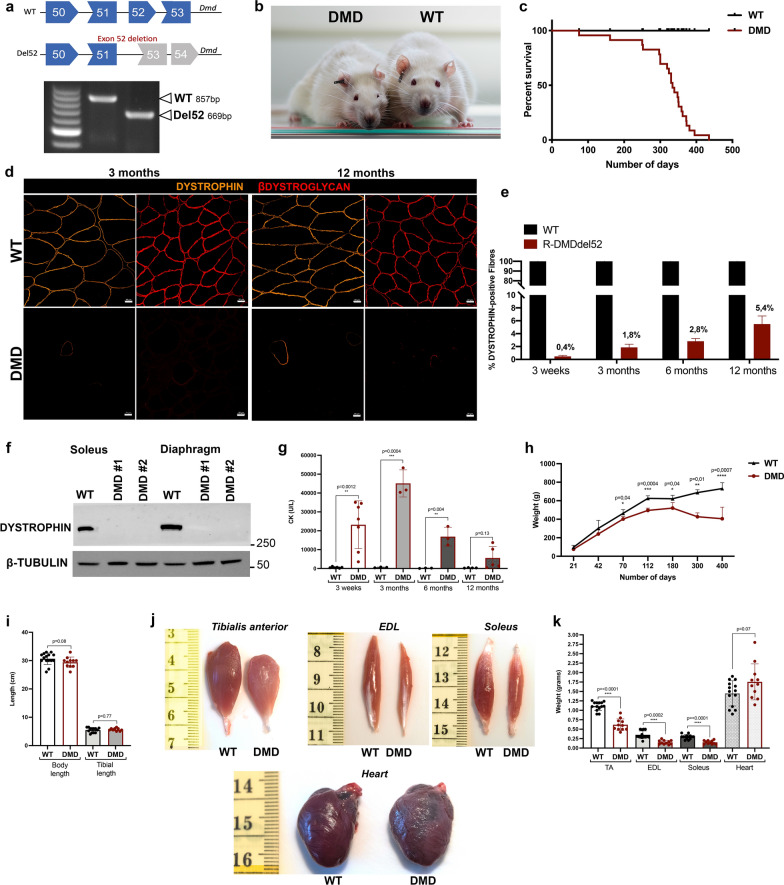
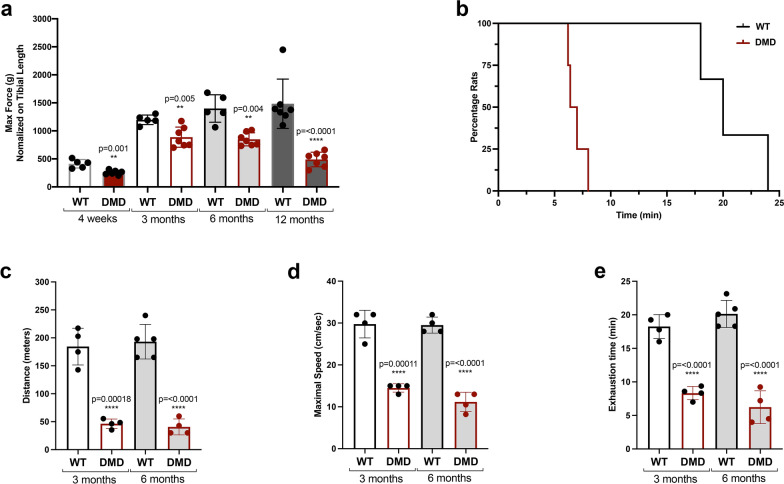
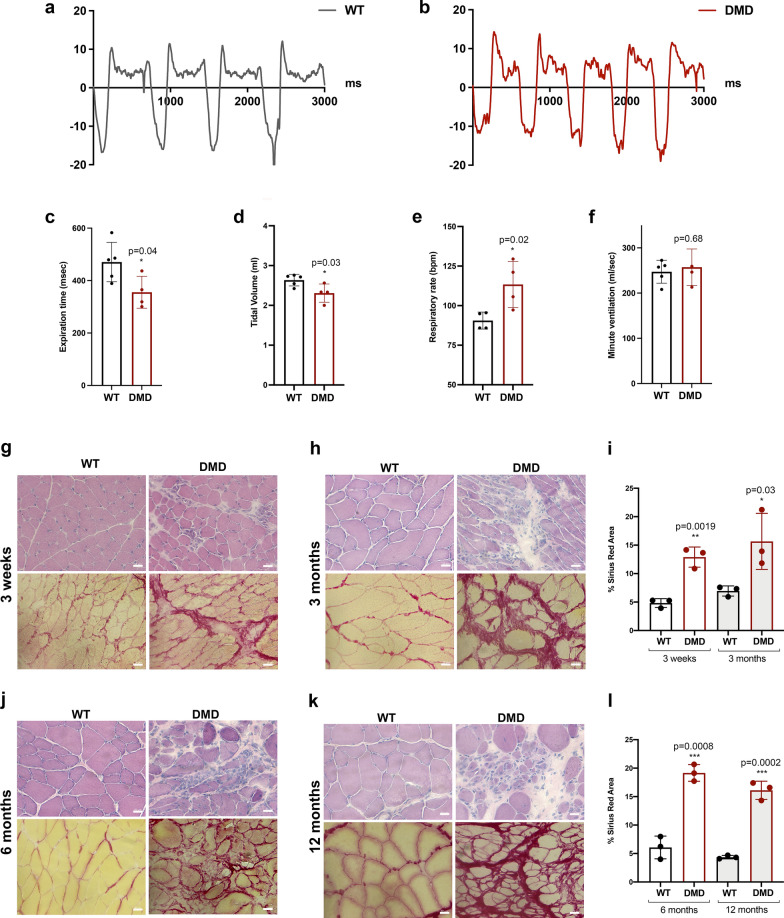
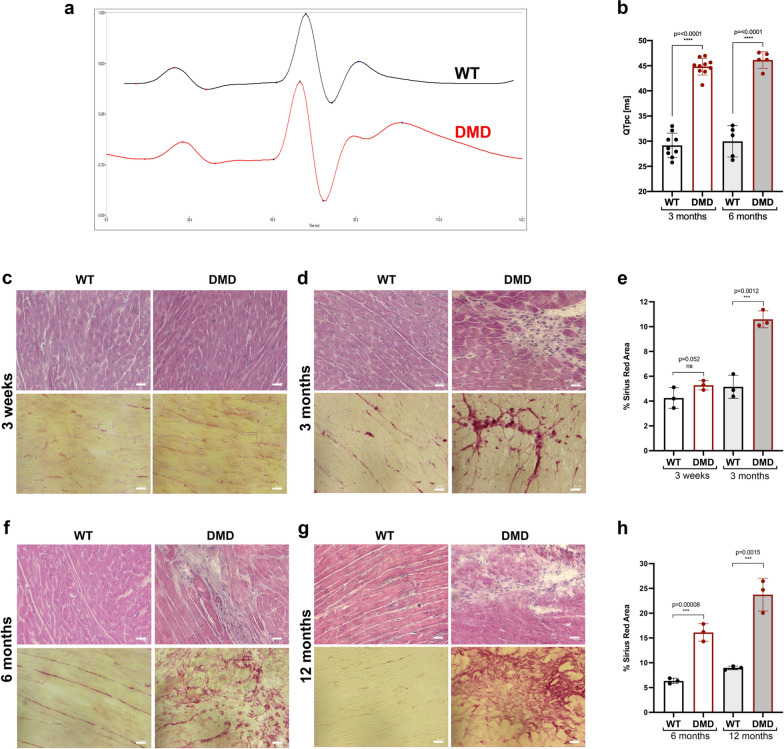
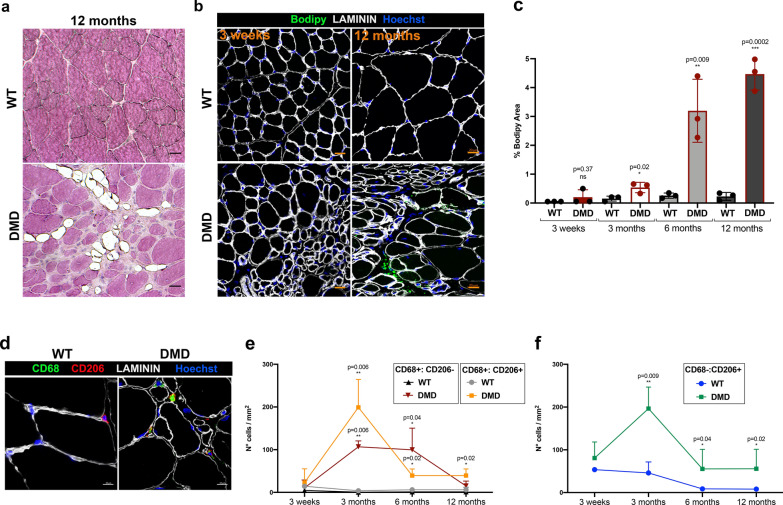
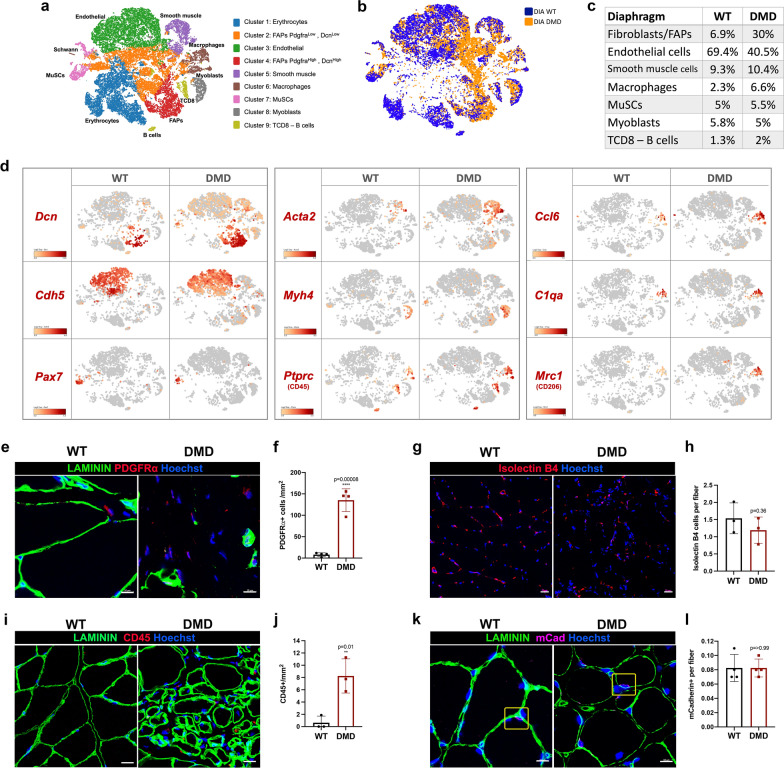
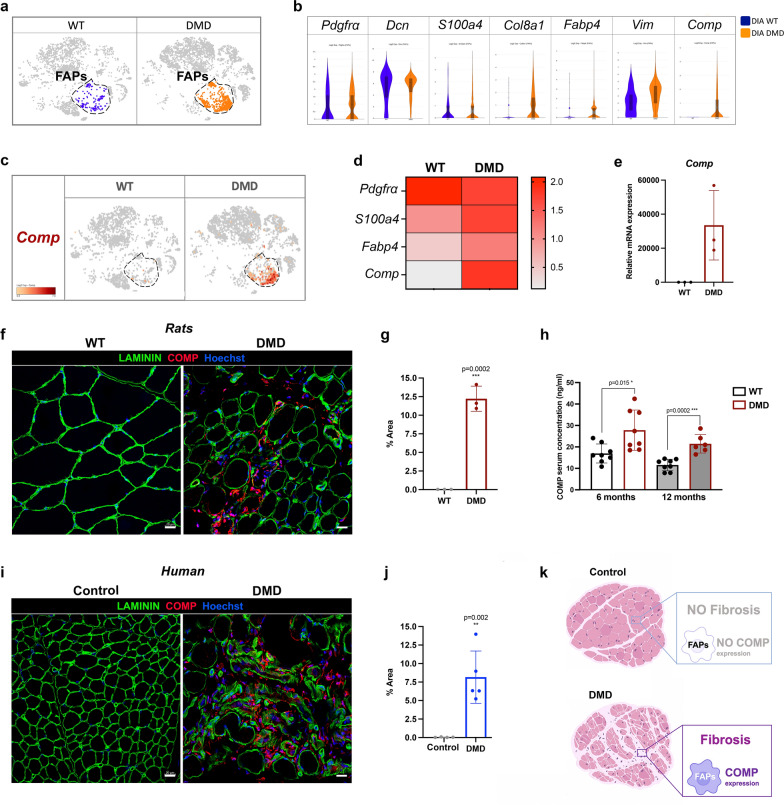
Duchenne muscular dystrophy (DMD) is a fatal muscle-wasting disorder caused by mutations in the Dystrophin gene and for which there is currently no cure. To bridge the gap between preclinical and therapeutic evaluation studies, we have generated a rat model for DMD that carries an exon 52 deletion (R-DMDdel52) causing a complete lack of dystrophin protein. Here we show that R-DMDdel52 animals recapitulated human DMD pathophysiological trajectory more faithfully than the mdx mouse model. We report that R-DMDdel52 rats displayed progressive and severe skeletal muscle loss associated with fibrotic deposition, fat infiltration and fibre type switch. Early fibrosis was also apparent in the cardiac muscle. These histological modifications led to severe muscle, respiratory and cardiac functional impairments leading to premature death around 1 year. Moreover, DMD muscle exhibited systemic inflammation with a mixed M1/M2 phenotype. A comparative single cell RNAseq analysis of the diaphragm muscle was performed, revealing cellular populations alteration and molecular modifications in all muscle cell types. We show that DMD fibroadipogenic progenitors produced elevated levels of cartilage oligomeric matrix protein, a glycoprotein responsible for modulating homeostasis of extracellular matrix, and whose increased concentration correlated with muscle fibrosis both in R-DMDdel52 rats and human patients. Fibrosis is a component of tissue remodelling impacting the whole musculature of DMD patients, at the tissue level but most importantly at the functional level. We therefore propose that this specific biomarker can optimize the prognostic monitoring of functional improvement of patients included in clinical trials.
Keywords: Duchenne muscular dystrophy; Long QT; Preclinical modelling; Skeletal muscle; Translational medicine; scRNAseq.
© 2022. The Author(s).
Conflict of interest statement
The authors have no relevant financial or non-financial interests to disclose and no competing interests to declare that are relevant to the content of this article. All the procedures including animal handling were validated by the ethic committee of the French ministry (APAFIS#25606-202005311746599) and all the DMD patients and controls gave their informed consent after the explanation of the research studies.
Figures
References
-
- Acharya C, Yik JH, Kishore A, Van Dinh V, Di Cesare PE, Haudenschild DR. Cartilage oligomeric matrix protein and its binding partners in the cartilage extracellular matrix: interaction, regulation and role in chondrogenesis. Matrix Biol. 2014;37:102–111. doi: 10.1016/j.matbio.2014.06.001. - DOI - PubMed
-
- Agarwal P, Zwolanek D, Keene DR, Schulz JN, Blumbach K, Heinegard D, Zaucke F, Paulsson M, Krieg T, Koch M, et al. Collagen XII and XIV, new partners of cartilage oligomeric matrix protein in the skin extracellular matrix suprastructure. J Biol Chem. 2012;287:22549–22559. doi: 10.1074/jbc.M111.335935. - DOI - PMC - PubMed
-
- Aikawa T, Takeda A, Oyama-Manabe N, Naya M, Yamazawa H, Koyanagawa K, Ito YM, Anzai T. Progressive left ventricular dysfunction and myocardial fibrosis in Duchenne and Becker muscular dystrophy: a longitudinal cardiovascular magnetic resonance study. Pediatr Cardiol. 2019;40:384–392. doi: 10.1007/s00246-018-2046-x. - DOI - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
