

Plasma cell-free RNA profiling distinguishes cancers from pre-malignant conditions in solid and hematologic malignancies
- PMID: 35468987
- PMCID: PMC9038724
- DOI: 10.1038/s41698-022-00270-y
Plasma cell-free RNA profiling distinguishes cancers from pre-malignant conditions in solid and hematologic malignancies
Abstract
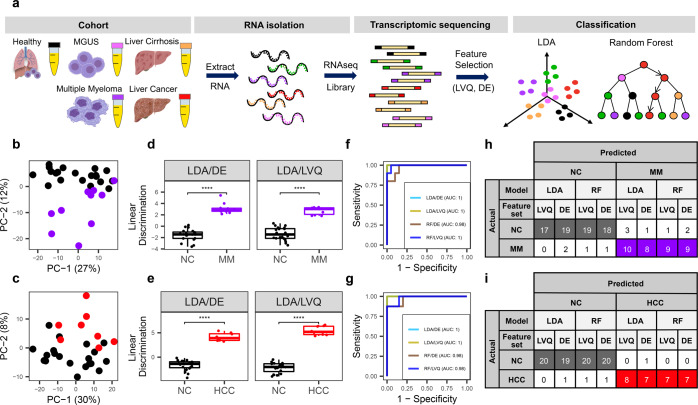
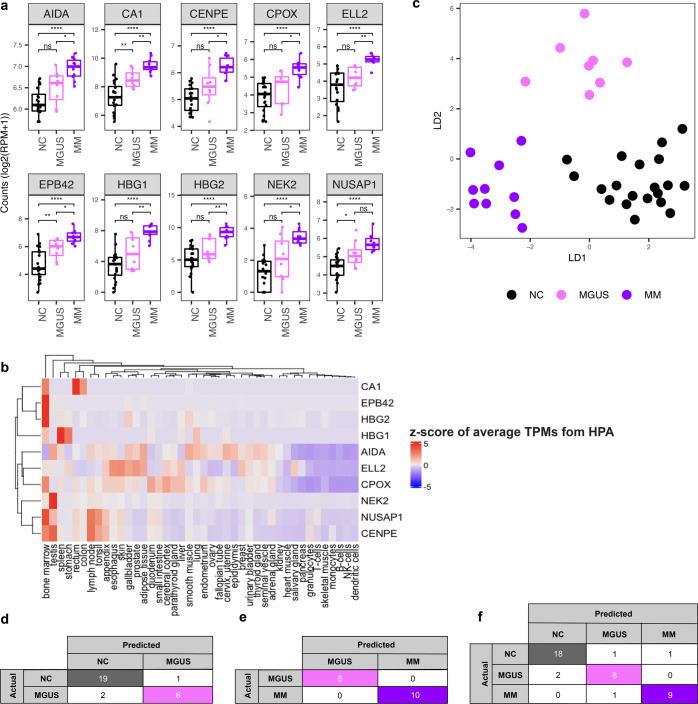
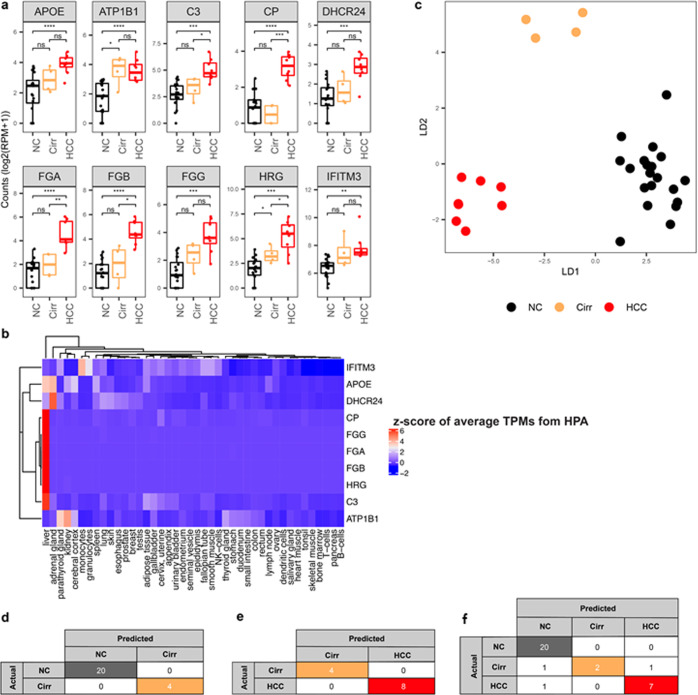
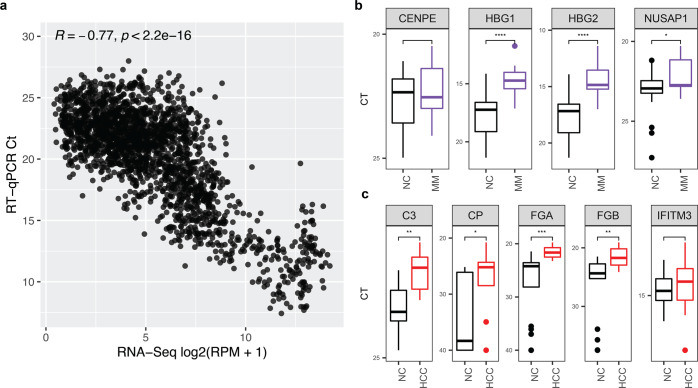
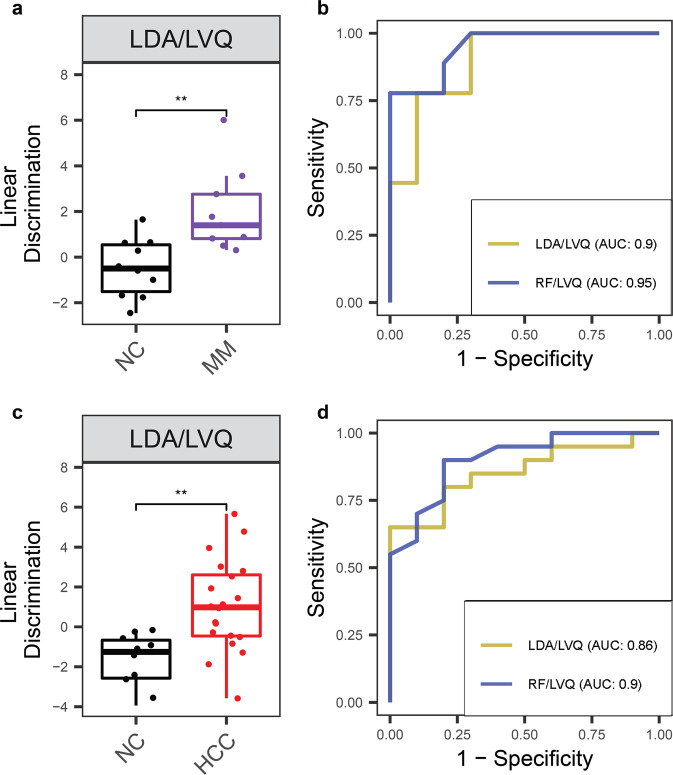
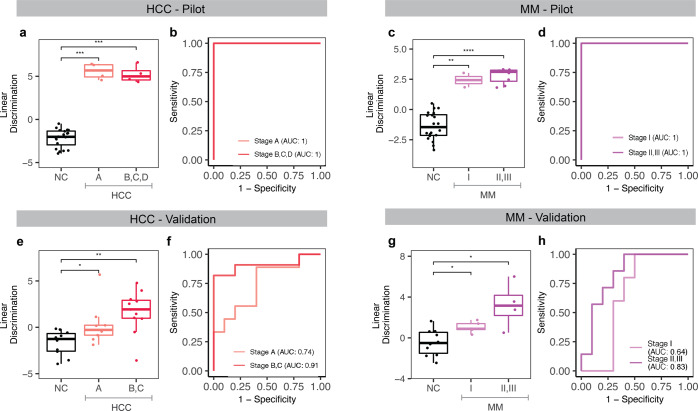
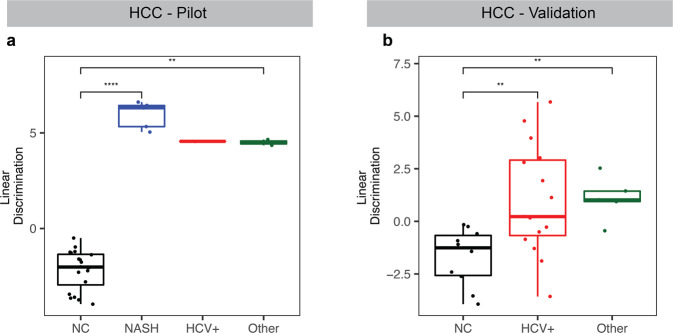
Cell-free RNA (cfRNA) in plasma reflects phenotypic alterations of both localized sites of cancer and the systemic host response. Here we report that cfRNA sequencing enables the discovery of messenger RNA (mRNA) biomarkers in plasma with the tissue of origin-specific to cancer types and precancerous conditions in both solid and hematologic malignancies. To explore the diagnostic potential of total cfRNA from blood, we sequenced plasma samples of eight hepatocellular carcinoma (HCC) and ten multiple myeloma (MM) patients, 12 patients of their respective precancerous conditions, and 20 non-cancer (NC) donors. We identified distinct gene sets and built classification models using Random Forest and linear discriminant analysis algorithms that could distinguish cancer patients from premalignant conditions and NC individuals with high accuracy. Plasma cfRNA biomarkers of HCC are liver-specific genes and biomarkers of MM are highly expressed in the bone marrow compared to other tissues and are related to cell cycle processes. The cfRNA level of these biomarkers displayed a gradual transition from noncancerous states through precancerous conditions and cancer. Sequencing data were cross-validated by quantitative reverse transcription PCR and cfRNA biomarkers were validated in an independent sample set (20 HCC, 9 MM, and 10 NC) with AUC greater than 0.86. cfRNA results observed in precancerous conditions require further validation. This work demonstrates a proof of principle for using mRNA transcripts in plasma with a small panel of genes to distinguish between cancers, noncancerous states, and precancerous conditions.
© 2022. The Author(s).
Conflict of interest statement
Oregon Health and Science University has filed patent applications based on this work. The authors declare no competing interests.
Figures
References
-
- SEER. Cancer Stat Facts: Liver and Intrahepatic Bile Duct Cancer (National Cancer Institute, 2018)
-
- Howlader N. et al. SEER Cancer Statistics Review, 1975–2018 (National Cancer Institute, 2021)
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
