

A bidirectional switch in the Shank3 phosphorylation state biases synapses toward up- or downscaling
- PMID: 35471151
- PMCID: PMC9084893
- DOI: 10.7554/eLife.74277
A bidirectional switch in the Shank3 phosphorylation state biases synapses toward up- or downscaling
Abstract
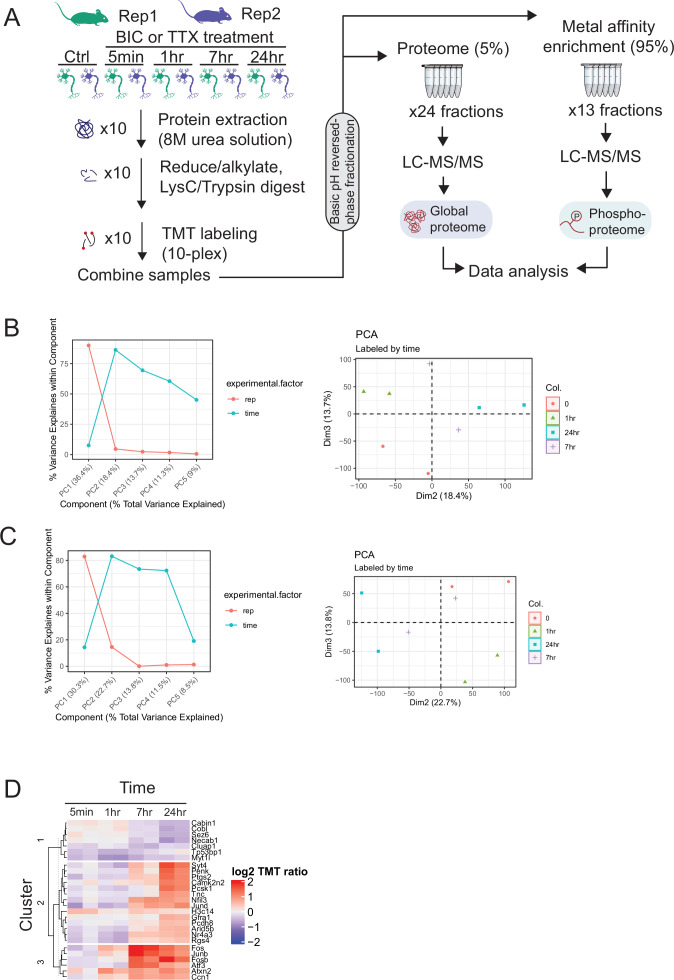
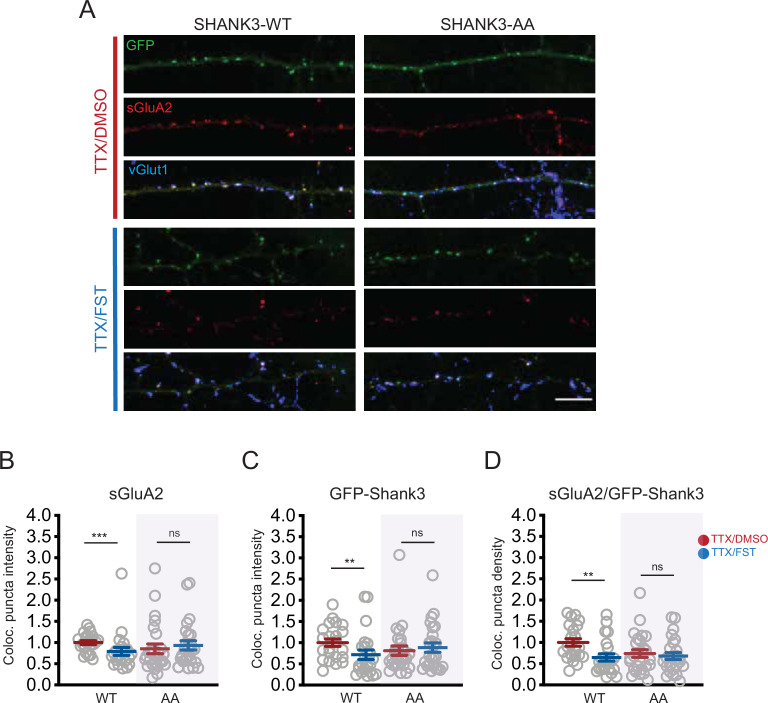
Homeostatic synaptic plasticity requires widespread remodeling of synaptic signaling and scaffolding networks, but the role of post-translational modifications in this process has not been systematically studied. Using deep-scale quantitative analysis of the phosphoproteome in mouse neocortical neurons, we found widespread and temporally complex changes during synaptic scaling up and down. We observed 424 bidirectionally modulated phosphosites that were strongly enriched for synapse-associated proteins, including S1539 in the autism spectrum disorder-associated synaptic scaffold protein Shank3. Using a parallel proteomic analysis performed on Shank3 isolated from rat neocortical neurons by immunoaffinity, we identified two sites that were persistently hypophosphorylated during scaling up and transiently hyperphosphorylated during scaling down: one (rat S1615) that corresponded to S1539 in mouse, and a second highly conserved site, rat S1586. The phosphorylation status of these sites modified the synaptic localization of Shank3 during scaling protocols, and dephosphorylation of these sites via PP2A activity was essential for the maintenance of synaptic scaling up. Finally, phosphomimetic mutations at these sites prevented scaling up but not down, while phosphodeficient mutations prevented scaling down but not up. These mutations did not impact baseline synaptic strength, indicating that they gate, rather than drive, the induction of synaptic scaling. Thus, an activity-dependent switch between hypo- and hyperphosphorylation at S1586 and S1615 of Shank3 enables scaling up or down, respectively. Collectively, our data show that activity-dependent phosphoproteome dynamics are important for the functional reconfiguration of synaptic scaffolds and can bias synapses toward upward or downward homeostatic plasticity.
Keywords: cell biology; homeostatic plasticity; mouse; neuroscience; phosphorylation; rat; synaptic scaling.
© 2022, Wu et al.
Conflict of interest statement
CW, VT, PJ, AG, HK, KK, MM, LL, SC, JC, GT No competing interests declared
Figures













References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
